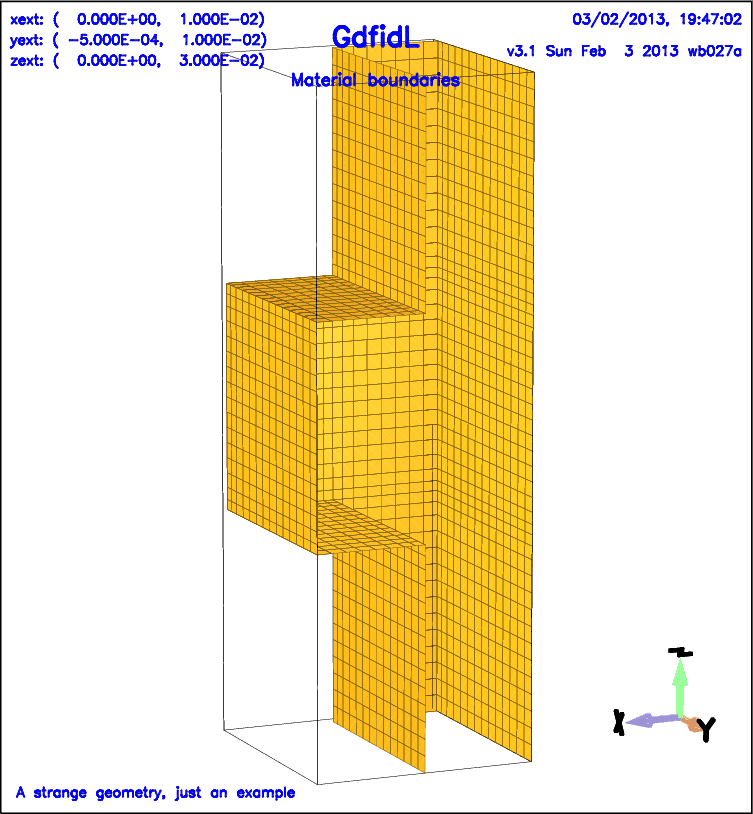
gd1 reads its information from the standard input unit. You will normally use two or more xterms to operate gd1. In one of the xterms you edit an inputfile that describes your geometry, in the other xterm you iteratively start gd1 and try out how gd1 reacts to your input.
gd1 reads the information about the geometry that you are interested in
from stdin or from a file that you specify via include(filename).
gd1 generates the mesh and computes the resonant fields or time dependent
fields, depending on the input you give it.
The results are written to a database that can be read by gd1.pp.
In the section -general, you specify the name of the file where the results shall be written to.
In the section -mesh, you have to specify the borderplanes of the computational volume, and the default mesh spacing. You also define the boundary conditions at the borderplanes here.
In the section -material, you specify the electric properties of the materials.
############################################################################## # Flags: nomenu, noprompt, nomessage, # ############################################################################## # gdfidl (64) (V 3.1) (compiled Fri Feb 1 18:37:42 GMT 2013 on Host wb032a) # # Vshzp 2013.02.01 # ############################################################################## # -general -- Output File, Annotations. # # -mesh -- Bounding Box, Spacing, fixed Meshplanes, # # -- Boundary Conditions. # # -material -- Material Properties. # # -lgeometry -- Load a previously used Geometry. # # ***** Geometric primitives **** # # -brick -- Simple rectangular Brick. # # -gccylinder -- Circular Cylinder in general Direction. # # -ggcylinder -- General Cylinder in general Direction. # # -gbor -- Body of Revolution in general Direction. # # -stlfile -- CAD Import via STL-File. # # -geofunction -- Analytic Function. # # -transform -- Rotations etc # # ***** Solver Sections **** # # -eigenvalues -- Resonant Fields # # -fdtd -- Time dependent Fields # # -magnetostatic -- Magnetostatic Fields (rudimentary) # # ******* # # -volumeplot -- Displays Mesh Filling. # # -cutplot -- Displays Mesh Filling in a single Plane # # ***** Miscellanea ****** # # -debug -- Specify Debug Levels # # # ############################################################################## # ?, end, help, __mdoit # ##############################################################################The menu shows all sections currently accessible. The section
-fdtd has some subsections.
You enter a section by specifying its name.
-general, you say:
-generalSince all commands may be abbreviated, you may also say:
-ge
############################################################################## # Flags: nomenu, noprompt, nomessage, # ############################################################################## # Section: -general # ############################################################################## # outfile = /tmp/--username--/--SomeDirectory--/Results # # scratchbase= /tmp/--username--/--SomeDirectory--/scratch # # restartfiles= -none- # # t1restartfiles= 1440 -- Minutes. When to write the first Set. # # dtrestartfiles= 1440 -- Minutes. Distance between writing. # # stopafterrestartfiles=1000000000 -- Stop after writing so often. # # text( 1)= ' ' # # text( 2)= ' ' # # text( 3)= ' ' # # text( 4)= ' ' # # text( 5)= ' ' # # text( 6)= ' ' # # text( 7)= ' ' # # text( 8)= ' ' # # text( 9)= ' ' # # text(10)= ' ' # # text(11)= ' ' # # text(12)= ' ' # # text(13)= ' ' # # text(14)= ' ' # # text(15)= ' ' # # text(16)= ' ' # # text(17)= ' ' # # text(18)= ' ' # # text(19)= ' ' # # text(20)= ' ' # # ndpw = auto -- PVM/MPI: Number of Dice per Worker # # iodice= no -- PVM : Each Worker writes its own Results. # # nrofthreads= 8 -- SMP and PVM/MPI: Nr of Threads per Worker. # # numa = yes -- SMP: Select NuMA-aware Schemes # # modnuma= 4 -- SMP: On What Cores to run. # ############################################################################## # ?, return, help # ##############################################################################
TMPDIR set,
gd1 will as default set scratchbase to the string
$TMPDIR/gdfidl-scratch-pid=XXXXX-
Here $TMPDIR is the value of the environment variable TMPDIR,
and XXXXX
is the numerical value of the process-id of gd1.
The size of the scratchfiles is often more than the amount of RAM that gd1 itself needs. The scratchfiles should be located on a disk which is directly attached to the compute nodes. If the scratchfiles are located on a network attached storage, much longer computation time is to be expected.
If you plan a cluster for using GdfidL, plane about ten times more directly attached scratch-space than installed RAM on each compute node.
restartfiles must be a valid filename in a
filesystem, which survives a system crash, ie. a filesystem which is not
cleared at boot time.
If a value for restartfiles is given,
the solver will write a set at selected times.
There will be up to two sets of restartfiles.
There are two sets, because according to murphy's law it will happen that a
system crash occurs just while one set is being written. In such a case,
the older set still can be used. When the restartfiles are written,
a comment is given about how to use the restartfiles.
Essentially this is: you have to restart the computation
on the same computer system (if using a parallel system: with the same
number of tasks, using the same nodes, using the same executable), with
an additional parameter on the command line of the master task.
The additional command line parameter is
-restartfiles=NAME-OF-SET .
Writing Restartfiles and Recovering works with PVM/MPI.
You can also use this facility to trick out batch systems. You can specify that restartfiles are to be written eg. after one hour, and the computation after writing the restartfiles shall stop. You then restart such a computation. This way, you can have long running GdfidL computations in a batch Queue which only accepts jobs with less than two hours wall clock time.
text()= ANY STRING, NOT NECESSARILY QUOTED
text(NUMBER)= ANY STRING, NOT NECESSARILY QUOTED ANY STRING, NOT NECESSARILY QUOTED:
NUMBER:
NUMBER, the string following
text()=
is placed in the next free line.
In the case with NUMBER, it is guaranteed, that the string
is placed in the NUMBER.st line.
You can specify up to 20 annotation strings,
the maximum length of each annotation string is 80 characters.
[auto|NUMBER]
When computing in parallel,
the computational volume is subdivided in NUMBER subvolumes per processor.
If ndpw=auto, the number of subvolumes (dice) is chosen such
that each subvolume contains about 1 million grid cells and the total
number of subvolumes is larger than the total number of threads to use.
The total number of threads to use is NROFTHREADS times the number of nodes
used for the parallel run.
[yes|no]
When computing in parallel,
the result of each subvolume is stored on the local disk of each
processor. This leads to faster writing of the results, but longer time
for postprocessing of the results.
This might be needed for very large parallel computations where the
computed electromagnetic fields must be loaded into the postprocessor
for computing, eg. the Q-value.
For very large parallel computations, it can happen that the computed
datasets are so large, that the postprocessor cannot handle the amount of
data in its limited address space (32 bit processors).
Even on 64 bit processors, the required memory might be larger than
what is available on the used system.
When iodice= yes, the results are written to the local
disks of the compute nodes. When the results shall be analysed by
gd1.pp, on each compute node an instance of gd1.pp will be started to handle
the local amount of data.
Only the PVM version of gd1.pp can be run in parallel. There is no MPI version of gd1.pp, as MPI lacks some needed functionality.
NTHREADS
The Number of Threads to use. Each Thread uses one CPU-Core.
[yes|no]
When numa= yes, Schemes that are optimized for Non-Uniform Memory Access
Machines are used.
MODNUMA
The Operating System is instructed that the i.th Thread shall run on one of the Cores
(i-1)*MODNUMA .. i*MODNUMA - 1
/tmp/UserName/restartfile.
Together with the plots that gd1.pp will produce,
the strings 'Parameter is 45' and '2*Parameter is 90' shall appear.
-general
outfile= /tmp/UserName/resultdirectory
scratch= /tmp/UserName/delete-me-
restartfiles= /tmp/UserName/restartfile
define(PARAMETER, 45)
text(1)= Parameter is PARAMETER
text(2)= 2*Parameter is eval(2*PARAMETER)
Example
Tell GdfidL that every XX Minutes, all Data describing the
current State of Computation shall be written to Files.
From these Data, a Computation can be restarted.
There is an Option, that after eg. two Datasets written, the Computation
shall stop.
1.2 A Script, which restarts as long as GdfidL has not yet signalled that
the Job is finished, is:
#!/bin/sh
#
# The Input for GdfidL shall contain a Specification
# for Restartfiles. In this Example, we do that
# via creating the './gdfidl.solver-profile', which is
# read and interpreted at the Start of Run.
(cat > ./gdfidl.solver-profile) << UntilThisMarker
-general
# Write a Set of Restartfiles every 60 Minutes.
restartfiles= /tmp/UserName/restartfiles # Where to write the Restartfiles
t1restart= 60 # When to write the first Set of Restartfiles
dtrestart= 60 # Wall Clock Time Difference between writing Restartfiles
stopafterrestart= 2 # Stop after writing the second Set of Restartfiles.
UntilThisMarker
#
# The initial Run.
# Use '>' instead of '| tee Logfile' to get the Returncode of GdfidL.
gd1 -DWhateverYourOptions=... \
< Inputfile > Logfile-restart
RETURNCODE=$?
echo RC ist $RETURNCODE
#
# While GdfidL signals that a Restart is useful, restart.
while [ "$RETURNCODE" -eq "1" ]
do
gd1 -restartfiles=/tmp/UserName/restartfiles.iMod-2 \
>> Logfile-restart
RETURNCODE=$?
echo RC ist $RETURNCODE
if [ "$RETURNCODE" -eq "0" ]; then
exit 0
fi
done
#
exit 0
#######
Writing Restartfiles is useful anyway.
If Restartfiles are written, one has the Chance to restart a
crashed Computation which crashed because of some Failure of the
Computersystem. To restart a Computation, the Restartfiles must be
accessible, of course. So the Restartfiles should not be written
to /tmp, or some other Filesystem which might be cleaned at System
Boot Time or so.
When writing Restartfiles for the possibility of recovering from some Failure of the System, you would not specify
-general, stopafterrestart= 2
The writing of the Restartfiles takes some Time, Minutes or so.
So the '-general, dtrestart= XX' Parameter should be significantly
larger than '1'. We suggest '-general, dtrestart= 24*60'.
That says: Write Restartfiles once per 24 Hours.
If such a Computation crashed, inspect the Logfile. You shall find Lines similar to:
############### # Restartfiles are to be written. ######################################################################### # A Set of Restartfiles has been written. # To restart this Computation, start with the same Command, # on the same Computer System, but with the additional Parameter # -restartfiles=/tmp/UserName/restartfiles.iMod-1 #########################################################################This '
start with the same Command, on the same Computer System,'
is for Safety. A Restart will work as long as the same Executables
are used, the same PVM/MPI-Parameters are used, and the Restartfiles
are accessible from every used Node.
############################################################################## # Flags: nomenu, noprompt, nomessage, # ############################################################################## # Section -mesh # ############################################################################## ## Bounding Box: # # pxlow = undefined , pylow = undefined , pzlow = undefined # # pxhigh= undefined , pyhigh= undefined , pzhigh= undefined # # volume= (undefined, undefined, undefined, undefined, undefined, undefined)# # __zlowref = undefined # # __zhighref= undefined # ## Boundary Conditions: # # cxlow = electric, cylow = electric, czlow = electric # # cxhigh= electric, cyhigh= electric, czhigh= electric # ## Periodic BC's for loss-free Eigenvalues: # # xperiodic= no , xphase= undefined # # yperiodic= no , yphase= undefined # # zperiodic= no , zphase= undefined # # # # # # # # xspacing= undefined, xminspacing= undefined # # yspacing= undefined, yminspacing= undefined # # zspacing= undefined, zminspacing= undefined # # graded = no # # xgraded= no , ygraded= no , zgraded= no # # xqfgraded= 1.20, xdmaxgraded= undefined # # yqfgraded= 1.20, ydmaxgraded= undefined # # zqfgraded= 1.20, zdmaxgraded= undefined # # perfectmesh= yes # # geoscale= 1.0 # ## Commands: # # # # # # # # # # # # xfixed(N, X0, X1) -- N fixed Meshplanes between X0 and X1 # # yfixed(N, Y0, Y1) -- N fixed Meshplanes between Y0 and Y1 # # zfixed(N, Z0, Z1) -- N fixed Meshplanes between Z0 and Z1 # ############################################################################## # listplanes, return, help # ##############################################################################
__zlowref and above __zhighref
is translational invariant.
electric, magnetic.
yes, no.
The parameter toggle the application of periodic boundary conditions
between the lower and upper boundary planes in x-, y- or z-direction.
You can compute with more than one ?periodic= yes.
This is only implemented for eigenvalue computations.
For time-domain computation, these parameters should be set to no.
bricks, and where you enforce them via
xfixed, yfixed, zfixed,
if the distance between such fixed meshplanes is more than
minspacing.
Otherwise, one of these fixed meshplanes is deleted.
The deleted one is the meshplane with the higher coordinate value.
If this parameter minspacing is not given, gd1 uses the value of spacing/10 instead.
yes, no.
graded= yes, the space between fixed meshplanes will be filled
with a graded mesh. The ratio of adjacent mesh-spacing will be approximately
the value given for qfgraded.
The largest mesh-spacing will be less or equal dmaxgraded.
graded, but only that the grid planes in x-, y- or z-constant
are considered for grading.
graded= no).
It is only guaranteed, that the borders of bricks
lie on meshplanes.
If you have a geometry with items other
than bricks you can improve the mesh by enforcing meshplanes with
these commands.
xfixed(NUMBER, LOW, HIGH)
yfixed(NUMBER, LOW, HIGH)
zfixed(NUMBER, LOW, HIGH)
NUMBER:
LOW, HIGH.
The total number of meshplanes enforced
by such a command is exactly NUMBER.
In the case of xfixed,
the meshplanes are positioned at positions 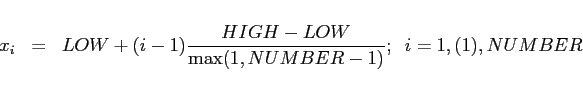
LOW, HIGH:
NUMBER NUMBER = 1, in this case
a single meshplane at LOW is enforced.
LOW is really lower
than HIGH.
It is normally counterproductive to specify regions of fixed meshplanes. Better only specify selected meshplanes, eg xfixed(2, X0, X1), or xfixed(1, X0, X0). Let the solver compute on a as homogeneous as possible mesh.
-mesh
spacing= 1e-3
pxlow= 1e-2, pxhigh= 0
pylow= 2e-2, pyhigh= 0
pzlow= 3e-2, pzhigh= 0
cxlow= ele, cxhigh= mag
cylow= ele, cyhigh= mag
zperiodic= yes, zphase= 120
type= electric),
perfect magnetic conducting (type= magnetic),
or may be loss-free or lossy dielectrics with anisotropic values for
type= normal).
Dielectrics may have up to 10 LORENTZ-resonances in their permittivity
and permeability functions.
############################################################################## # Flags: nomenu, noprompt, nomessage, # ############################################################################## # section -material # ############################################################################## # material= 3 epsr = undefined kappa = 0.0 # # type= undefined xepsr= undefined xkappa = 0.0 # # yepsr= undefined ykappa = 0.0 # # zepsr= undefined zkappa = 0.0 # # muer = undefined mkappa = 0.0 # # xmuer= undefined xmkappa= 0.0 # # ymuer= undefined ymkappa= 0.0 # # zmuer= undefined zmkappa= 0.0 # # # Dispersion Parameters. # # feps(1) = undefined fmue(1) = undefined # # xfeps(1)= undefined xfmue(1)= undefined # # yfeps(1)= undefined yfmue(1)= undefined # # zfeps(1)= undefined zfmue(1)= undefined # # aeps(1) = undefined amue(1) = undefined # # xaeps(1)= undefined xamue(1)= undefined # # yaeps(1)= undefined yamue(1)= undefined # # zaeps(1)= undefined zamue(1)= undefined # # fegm(1) = undefined fmgm(1) = undefined # # xfegm(1)= undefined xfmgm(1)= undefined # # yfegm(1)= undefined yfmgm(1)= undefined # # zfegm(1)= undefined zfmgm(1)= undefined # ############################################################################## # flow = undefined -- Plot feps(f) from this f # # fhigh = undefined -- upto this f. # # plotopts = -geometry 690x560+10+10 # # showtext = yes -- (yes | no) # # onlyplotfiles= no -- (yes | no) # ############################################################################## # return, showfeps, help # ##############################################################################
electric", "magnetic", "normal".
An "electric" material is treated as perfect electric conducting
for the field computation and a "magnetic" material is treated as
perfect magnetic conducting in the field computation.
lossy= no
in the section -eigenvalues is selected,
only the "epsr"
and "muer" of a "normal" material are used for the
field computation, ie. no losses, and no dispersive parameters
are modeled for eigenvalue computations.
The parameters "kappa" and "mkappa" of
materials with "type= normal" may be used by the
postprocessor,
gd1.pp,
to compute dielectric losses via a perturbation formula.
lossy= yes
in the section -eigenvalues is selected,
losses due to finite electric and magnetic conductivities,
and the dispersive parameters are taken into account.
epsr", "muer",
"kappa" and "mkappa"
as well as the dispersive parameters of a "type= normal"
material are used for the field computation.
type= electric" are considered perfectly conducting, and
materials with "type= magnetic" are considered perfectly
magnetic conducting.
Any specified electric conductivities for materials with
"type= electric" are used in the postprocessor,
gd1.pp,
to compute wall losses via a perturbation formula.
epsr= 3,
all three epsr values xepsr, yepsr and zepsr
are set to the value 3.
epsr matrices can be specified.
muer= 4,
all three muer values xmuer, ymuer and zmuer
are set to the value 4.
muer matrices can be specified.
kappa= 5,
all three kappa values xkappa, ykappa and zkappa
are set to the value 5.
kappa matrices can be specified.
mkappa= 6,
all three kappa values xmkappa, ymkappa
and zmkappa
are set to the value 6.
mkappa matrices can be specified.
feps(1)= 3e9,
all three feps(1) values xfeps(1), yfeps(1)
and zfeps(1)
are set to the value 3 GHz.
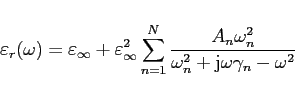 |
(1.1) |
| (1.2) | |||
| (1.3) | |||
| (1.4) | |||
| (1.5) |
epsr and
muer are 1. This is vacuum.
You can change the parameters epsr, muer, kappa
and mkappa of the
material '0', but you cannot change its type= normal.
type= electric.
type= magnetic.
-material
material= 3
type= magnetic
material= 4
type= normal, epsr= 3, kappa= 1, muer= 1
-material
# Dispersive Materials and Losses.
# To have Re(epsr) about 10 and Im(epsr) about 1 in 10-40GHz:
define(EPSQ, 10**2)
material= 3, type= normal, epsr= 10, muer= 1, kappa= 1
kappa= 0
define(i, 0)
define(i, i+1) feps(i)= 10e9, aeps(i)= 1.1/EPSQ, fegm(i)= 100e9
define(i, i+1) feps(i)= 20e9, aeps(i)= 0.42/EPSQ, fegm(i)= 100e9
define(i, i+1) feps(i)= 30e9, aeps(i)= 0.26/EPSQ, fegm(i)= 100e9
define(i, i+1) feps(i)= 40e9, aeps(i)= 0.28/EPSQ, fegm(i)= 100e9
flow= 1e9, fhigh= 70e9, showfeps
To compute an example of dispersive Materials:
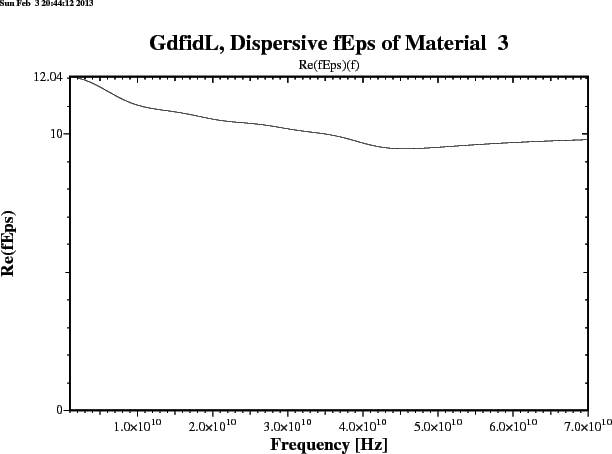
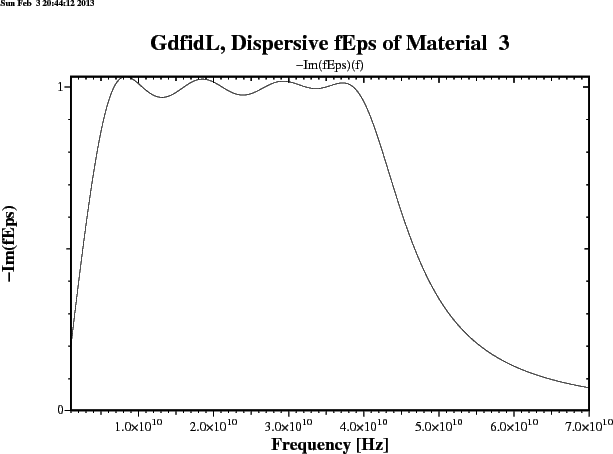
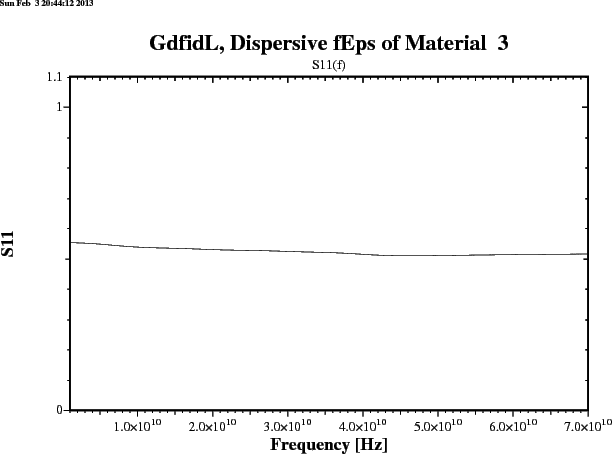
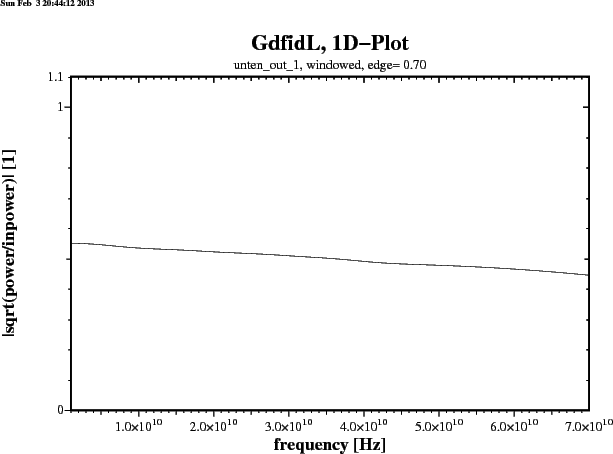
gd1 < /usr/local/gd1/examples-from-the-manual/dispersive-TEM.gdf | tee logfileWe get three plots. Figure 1.1 shows the real part, figure 1.2 shows the imaginary part of the frequency dependent epsr(f). Figure 1.3 shows the reflection that an infinitely large plane of such a material would produce for a perpendicular incident TEM-wave. The numerically computed reflection of a thick slab of such a material is given in figure 1.4.
############################################################################## # Flags: nomenu, noprompt, nomessage, # ############################################################################## # Section: -lgeometry # ############################################################################## # infile = -none- # # # # # ############################################################################## # ?, doit, return, end, help # ##############################################################################
infile= NAME_OF_A_RESULTFILE:
outfile of a previous computation.
doit:
-lgeometry, all previous defined geometric
items are lost.
You should only redefine material parameters after loading a grid via
-lgeometry.
brick is a rectangular box, with its edges parallel
to the cartesian coordinate axes.
If you need to model a brick with edges in other directions, you can
use the -rotate section to rotate the brick in any direction.
############################################################################## # Flags: nomenu, noprompt, nomessage, # ############################################################################## # section -brick # ############################################################################## # material= 1, sloppy= no # # whichcells= all, taboo= none # # show= off -- [off|now|later|all] # # name= brick-000000000 # # xlow = undefined , ylow = undefined , zlow = undefined # # xhigh= undefined , yhigh= undefined , zhigh= undefined # # # # # # # # volume= (undefined, undefined, undefined, undefined, undefined, undefined) # ############################################################################## # doit, return, help # ##############################################################################
yes, no. If no, meshplanes are enforced
at the borderplanes of the brick.
For a brick which is rotated, sloppy=no might give unexpected results.
all, or a material-index.
If whichcells= all, all volume inside the brick is assigned to
the material-index, provided the former material is not
taboo.
If whichcells is a material-index, only the parts of the
brick that are currently filled with the given index are assigned
the new material-index.
none, or a material-index.
If taboo= all, all volume inside the brick is assigned
the material-index.
If taboo is a material-index, only the parts of the
brick that are currently filled with another index than the given
index are assigned the new material-index.
show=off, no outline will be displayed.
show=later,
the outline of the brick will be shown later, together
with outlines of other specified items.
If show=all is present,
the outlines of all other specified items bricks
gccylinders, ggcylinders and gbors
found in the inputstream so far where show
was not off will be displayed.
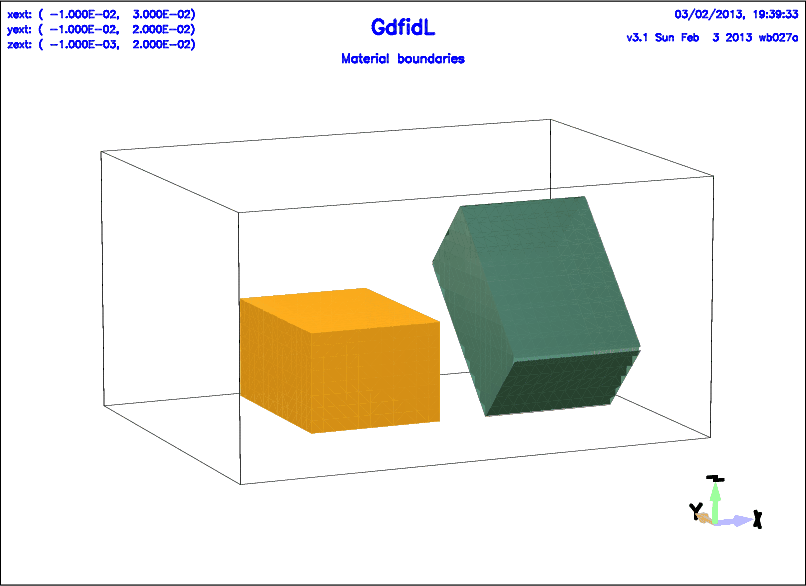
# /usr/local/gd1/examples-from-the-manual/brick-example.gdf
-general
outfile= /tmp/UserName/example
scratch= /tmp/UserName/scratch
-mesh
pxlow= -1e-2, pxhigh= 3e-2
pylow= -1e-2, pyhigh= 2e-2
pzlow= -0.1e-2, pzhigh= 2e-2
cxlow= ele, cxhigh= mag
cylow= ele, cyhigh= mag
czlow= ele, czhigh= mag
spacing= 1e-3
-brick
material= 1, sloppy= no
xlow= 0, xhigh= 1.1e-2
ylow= 0, yhigh= 1.5e-2
zlow= 0, zhigh= 0.8e-2
doit
-transform,
-translate, offset= ( 1.5e-2, 0, 0 ), doit
-rotate, axis= ( 1, 0, 0 ), angle= -45, doit
-brick, material= 3, sloppy= yes, doit
-volumeplot
scale= 3
doit
gccylinder is a circular cylinder, with its axis in
an arbitrary direction.
############################################################################## # Flags: nomenu, noprompt, nomessage, # ############################################################################## # section -gccylinder # ############################################################################## # material = 1 # # whichcells= all, taboo= none # # show= off -- [off|now|later|all] # # name= gccyl-000000000 # # radius = undefined # # length = undefined # # origin = ( undefined, undefined, undefined ) # # direction = ( undefined, undefined, undefined ) # ############################################################################## # doit, return, help # ##############################################################################
gccylinder.
all, or a material-index.
If whichcells=all, all volume inside the cylinder is assigned
the material-index, provided the former material is not
taboo.
If whichcells is a material-index, only the parts of the
cylinder that are currently filled with the given index are assigned
the new material-index.
none, or a material-index.
If taboo=none, all volume inside the cylinder is assigned
the material-index.
If taboo is a material-index, only the parts of the
cylinder that are currently filled with another index than the given
index are assigned the new material-index.
show=off, no outline will be displayed.
show=later,
the outline of the gccylinder will be shown later, together
with outlines of other specified items.
If show=all is present,
the outlines of all other specified items bricks
gccylinders, ggcylinders and gbors
found in the inputstream so far where show
was not off will be displayed.
-gbor, see pages ![[*]](crossref.gif) ff.
ff.
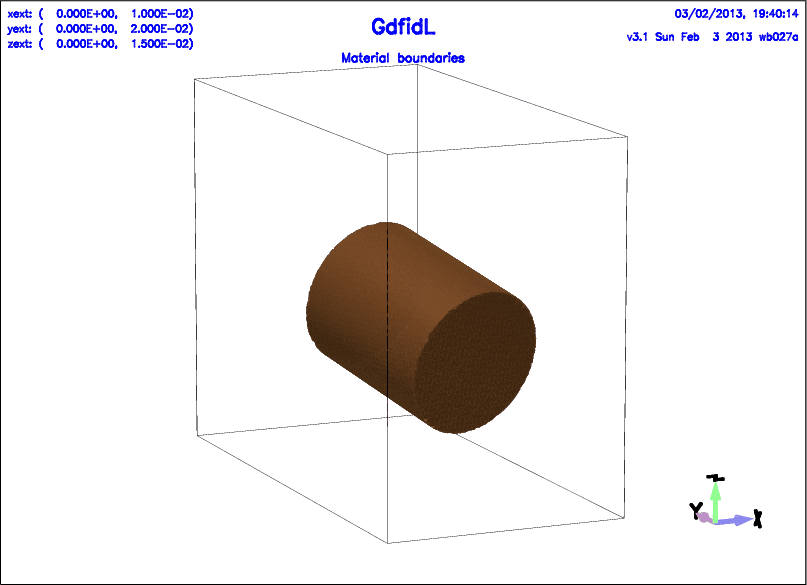
# /usr/local/gd1/examples-from-the-manual/gccylinder-example.gdf
-general
outfile= /tmp/UserName/example
scratch= /tmp/UserName/scratch-
-mesh
pxlow= 0, pxhigh= 1e-2
pylow= 0, pyhigh= 2e-2
pzlow= 0, pzhigh= 1.5e-2
cxlow= ele, cxhigh= mag
cylow= ele, cyhigh= mag
czlow= ele, czhigh= mag
spacing= 0.2e-3
-gccylinder
material= 5, radius= 3e-3, length= 7e-3
origin= ( 0.5e-2, 0.3e-2, 0.6e-2 )
direction= ( -0.4, 1.5, 0.4 )
doit
-volumeplot
scale= 3
doit
Example
# /usr/local/gd1/examples-from-the-manual/gccylinder-example2.gdf
define(VeryLarge, 10000)
-general
outfile= /tmp/UserName/example
scratch= /tmp/UserName/scratch
text()= A chain of circular cylinders
text()= The cylinders are partly outside of the bounding box.
-mesh
pxlow= -0.8, pxhigh= 10
pylow= -2, pyhigh= 2
pzlow= -0, pzhigh= 1.8
spacing= 8e-2
-brick
material= 0
volume= ( -VeryLarge, VeryLarge, \
-VeryLarge, VeryLarge, \
-VeryLarge, VeryLarge )
doit
define(RADIUS, 0.6)
-gccylinder
length= 1.7
radius= RADIUS
do jj= 0, 9, 1
material= jj+3
origin= ( jj*1.5*RADIUS, 0, 0 )
define(PHI, jj*22.5*@pi/180 )
direction= ( 0, cos(PHI), sin(PHI) )
doit
enddo
-volumeplot
eyeposition= ( 1.0, 2.30, 2 )
scale= 5
doit
 |
ggcylinder is a general cylinder with a footprint (cross-section)
described as a general polygon.
This footprint is swept along an axis in a general direction,
additionally, this footprint can shrink or expand along this axis,
additionally, this footprint can be rotated along the axis,
additionally, only parts of the ggcylinder that fulfill
some additional condition will be filled.
############################################################################## # Flags: nomenu, noprompt, nomessage, # ############################################################################## # section -ggcylinder # ############################################################################## # material = 1 # # whichcells = all, taboo= none # # show = off -- (off | all | later | now) # # name = ggcyl-000000000 # # fixpoints= no -- (yes|no) # # inside = yes -- (yes|no) # # originprime = ( 0.0, 0.0, 0.0 ) # # xprimedirection= ( 1.0, 0.0, 0.0 ) # # yprimedirection= ( 0.0, 1.0, 0.0 ) # # zprimedirection= ( 0.0, 0.0, 1.0 ) # # usezprimedirection= no -- (yes|no) # # range = ( undefined, undefined ) # # pitch = 0.0 -- [Degs/m] # # xexpgrowth = 0.0 # # yexpgrowth = 0.0 # # xslope = 0.0 -- (x2/x1-1)/len [1/m] # # yslope = 0.0 -- (y2/y1-1)/len [1/m] # # xscaleprime= 1.0 # # yscaleprime= 1.0 # # zxscaletablefile= -none- # zyscaletablefile= -none- # deltaphi= 4.0 -- Arc and Ellipse Resolution [Degs] # ############################################################################## ## Syntax: # # point= (Xi, Yi) # # arc, radius= RADIUS, type= [clockwise | counterclockwise] # # size= [small | large ] # # deltaphi= 5 # # ellipse, center= (X0, Y0), size= [small | large ] # # deltaphi= 5 # ############################################################################## # doit, return, help, list, reset, clear # ##############################################################################
material=MAT:
whichcells
all, or a material-index.
whichcells=all, all volume inside the
ggcylinder is assigned the material-index,
provided the former material is not taboo.
If whichcells is a material-index, only the parts of the
ggcylinder that are currently filled with the given index
are assigned the new material-index.
taboo
none, or a material-index.
taboo=none, all volume inside the
ggcylinder is assigned the material-index.
If taboo is a material-index, only the parts of the
ggcylinder that are currently filled with another index
than the given index are assigned the new material-index.
originprime:
ggcylinder.
xprimedirection:
yprimedirection:
usezprimedirection= [yes|no]:
usezprimedirection= yes, the axis of the cylinder is not
computed from the cross product of xprimedirection and yprimedirection,
but is taken to be the direction given by
zprimedirection= ( XZ, YZ, ZZ ).
zprimedirection= ( XZ, YZ, ZZ ):
usezprimedirection= yes, the axis of the cylinder has the
direction given by zprimedirection= ( XZ, YZ, ZZ ).
If usezprimedirection= no, the given values are not used
and the direction is given by the cross product of
xprimedirection and yprimedirection.
range:
xprimedirection,yprimedirection,(xprime
yprime), relative to
originprime.
pitch:
xexpgrowth, yexpgrowth
xslope, yslope:
xscaleprime, yscaleprime:
zxscaletablefile, zyscaletablefile:
inside:
ggcylinder
should be assigned material index MAT, or
whether the cells outside of it should be changed.
show:
show=off, no outline will be displayed.
show=later,
the outline of the ggcylinder will be shown later, together
with outlines of other specified items.
If show=all is present,
the outlines of all other specified items bricks
gccylinders, ggcylinders and gbors
found in the inputstream so far where show
was not off will be displayed.
fixpoints
point= (XI, YI):
XI, YI are the coordinates of the i.th point in the polygon
that describes the footprint of the ggcylinder.
There have to be minimum 3 points,
or 2 points and an arc
or 2 points and an ellipse.
arc:
radius= RADIUS:
RADIUS
size= [small | large] (optional):
type= [clockwise | counterclockwise]:
clear:
doit:
Example
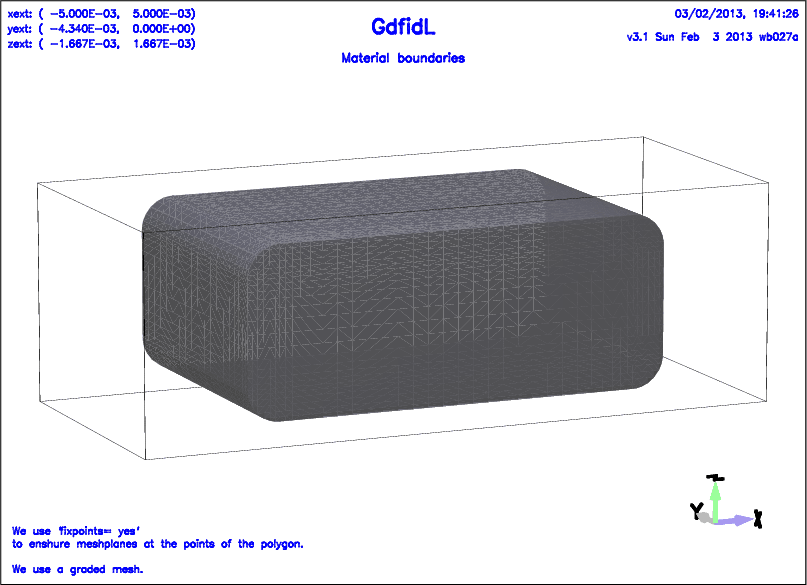
The following decribes a cavity with rounded corners.
# /usr/local/gd1/examples-from-the-manual/ggcylinder-example.gdf
-general
outfile= /tmp/UserName/example
scratch= /tmp/UserName/scratch
text()= We use 'fixpoints= yes'
text()= to enshure meshplanes at the points of the polygon.
text()=
text()= We use a graded mesh.
-mesh
spacing= 100.0e-06
graded= yes, dmaxgraded= 263.3e-06
pxlow= -5e-03, pxhigh= 5e-03
pylow= -4.34e-03, pyhigh= 0
pzlow= -1.6665e-03, pzhigh= 1.6665e-03
cxlow= ele, cxhigh= mag
cylow= ele, cyhigh= mag
czlow= ele, czhigh= ele
-ggcylinder
material= 7
originprime= ( 0, 0, 0 )
xprimedirection= ( 1, 0, 0 )
yprimedirection= ( 0, 0, 1 )
range= ( -4.2e-03, 4.2e-03 )
clear # clear the polygon-list, if any
point= ( -3.3405e-03, -816.5e-06 )
arc, radius= 500.0e-06, type= counterclockwise, size= small
point= ( -2.8405e-03, -1.3165e-03 )
point= ( 2.8405e-03, -1.3165e-03 )
arc
point= ( 3.3405e-03, -816.5e-06 )
point= ( 3.3405e-03, 816.5e-06 )
arc
point= ( 2.8405e-03, 1.3165e-03 )
point= ( -2.8405e-03, 1.3165e-03 )
arc
point= ( -3.3405e-03, 816.5e-06 )
fixpoints= yes # enshure mesh-planes at the points of the polygon
doit
-volumeplot
scale= 4
doit
Example
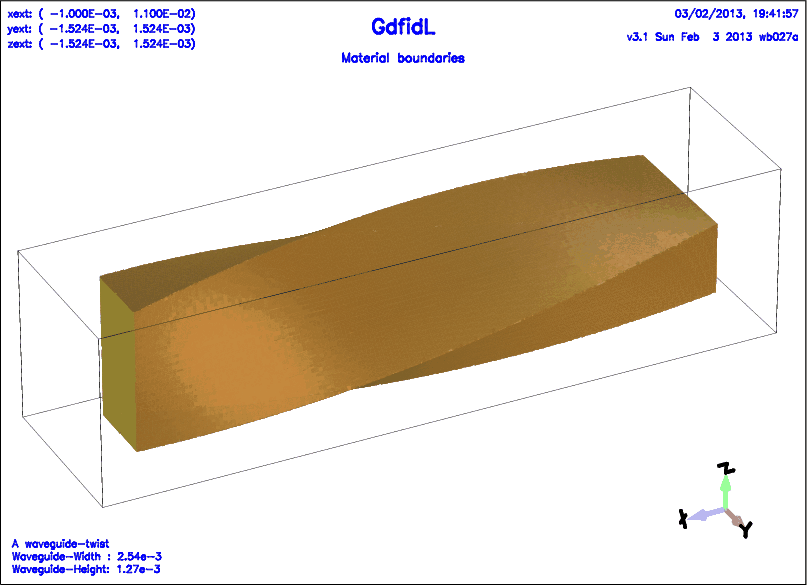
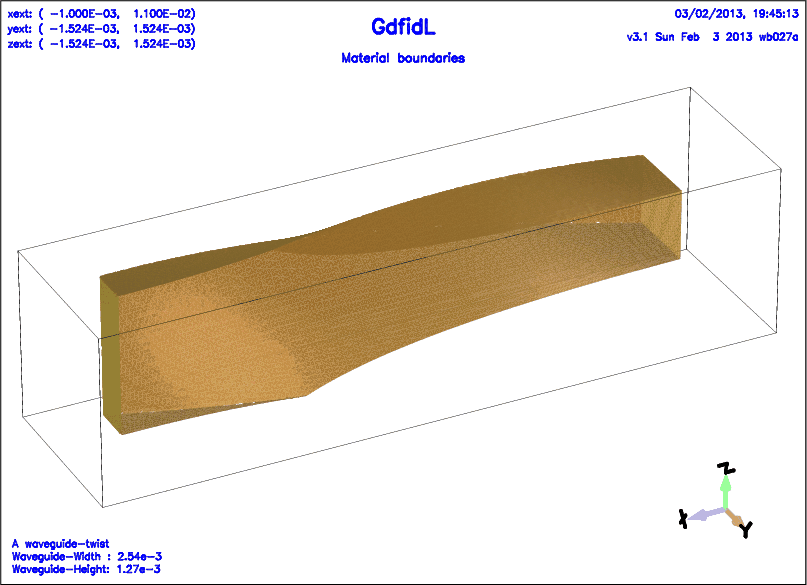
The following decribes a twisted rectangular waveguide.
# /usr/local/gd1/examples-from-the-manual/ggcylinder-twisted.gdf
define(LargeNumber, 10000)
define(LENGTH, 10e-3)
define(WGW, 2.54e-3 )
define(WGH, WGW/2 )
-general
outfile= /tmp/UserName/example
scratch= /tmp/UserName/scratch
text()= A waveguide-twist
text()= Waveguide-Width : WGW
text()= Waveguide-Height: WGH
-mesh
spacing= WGW/40
pxlow= -1e-3, pxhigh= LENGTH+1e-3
pylow= -WGW*0.6, pyhigh= 0.6*WGW
pzlow= -WGW*0.6, pzhigh= 0.6*WGW
define(EL, 10)
-material, material= EL, type= electric
-brick
#
# Fill the universe with metal:
#
material= EL
volume= ( -LargeNumber, LargeNumber, \
-LargeNumber, LargeNumber, \
-LargeNumber, LargeNumber )
doit
-ggcylinder
#
# The twisted waveguide.
# We use a rectangular footprint,
# and specify a pitch.
# The footprint shall rotate by -90 degrees, over a length of LENGTH.
#
material= 0
originprime= ( 0, 0, 0 )
xprimedirection= ( 0, 1, 0 )
yprimedirection= ( 0, 0, 1 )
range= ( 0, LENGTH )
pitch= -90/LENGTH
clear # clear the previous polygon-list, if any
point= ( -WGW/2, -WGH/2 )
point= ( WGW/2, -WGH/2 )
point= ( WGW/2, WGH/2 )
point= ( -WGW/2, WGH/2 )
doit
-volumeplot
eyeposition= ( 1, 2, 1.3 )
scale= 4.5
doit
Example
The following decribes a transition from a circular waveguide to
an elliptical waveguide.
# /usr/local/gd1/examples-from-the-manual/ggcylinder-circular-to-elliptic.gdf
define(LargeNumber, 10000)
define(LENGTH, 10e-3)
define(RADIUS1, 2.54e-3 ) define(RADIUS2, 5.0e-3 )
-general
outfile= /tmp/UserName/example
scratch= /tmp/UserName/scratch
text()= A transition from a circular waveguide to an elliptical one.
text()= The bounding box is specified such,
text()= that only the part below the plane z=0 is discretised.
-mesh
spacing= RADIUS1/20
pxlow= -1e-3, pxhigh= LENGTH+1e-3
pylow= -RADIUS2*1.0, pyhigh= RADIUS2*1.0
pzlow= -RADIUS1*1.1, pzhigh= 0
define(EL, 10)
-material, material= EL, type= electric
-brick
#
# Fill the universe with metal:
#
material= EL
volume= ( -LargeNumber, LargeNumber, \
-LargeNumber, LargeNumber, \
-LargeNumber, LargeNumber )
doit
-ggcylinder
#
# The waveguide.
# We use a circular footprint,
# and specify a slope, different in x- and y
#
material= 0
originprime= ( 0, 0, 0 )
xprimedirection= ( 0, 1, 0 )
yprimedirection= ( 0, 0, 1 )
range= ( 0, LENGTH )
# xlingro 1+(RADIUS2/RADIUS1-1)/LENGTH
xslope= (RADIUS2/RADIUS1-1)/LENGTH
yslope= 0
clear # clear the previous polygon-list, if any
point= ( -RADIUS1, 0 )
arc, radius= RADIUS1, size= large, type= counterclockwise
point= ( RADIUS1, 0 )
arc
point= ( -RADIUS1, 0 )
doit
-volumeplot
eyeposition= ( 2, 1, 1.8 )
scale= 3
doit
Example
The following decribes a elliptical wedge where the axis of the ggcylinder
is in the z-direction, while the plane normal of the wedge is titled.
# /usr/local/gd1/examples-from-the-manual/ggcylinder-usezprimedirection.gdf
#
# Some helpful symbols:
#
define(EL, 1) define(MAG, 2)
define(INF, 1000)
define(STPSZE, 0.5e-3) # define the transverse mesh step size, 0.5 mm
#
# description of the flange geometry
#
# define(FlangeGap, 2e-3 ) # gap of the flange joint
define(FlangeGap, 10e-3 ) # gap of the flange joint
define(FlangeD0, 80e-3) # diameter of gasket seal
define(Width, FlangeGap/2 ) # 1/2 width of flange gap in z-axis
define(FlangeRadius, FlangeD0/2 )
define(AxisA , 70e-3/2 ) # 1/2 *major diameter of interior ellipse
define(AxisB , 32e-3/2 ) # 1/2 *minor diameter of interior ellipse
#--- parameters related to a tilted flange joint ---
define(ZW, 10e-3) # z-deviation from the y-axis
define(Theta, atan(ZW/FlangeRadius) ) # angle between y-axis and y'-axis
define(RR, FlangeRadius/cos(Theta) ) # radius of tilted cylinder
define(LL, Width/cos(Theta) ) # half length of tilted cylinder
###
### We enter the section "-general"
### Here we define the name of the database where the
### results of the computation shall be written to.
### (outfile= )
### We also define what names shall be used for scratchfiles.
### (scratchbase= )
###
-general
outfile= /tmp/UserName/bla
scratch= /tmp/UserName/scratch-
text()= flange gap= FlangeGap
text()= mesh= STPSZE m
text()= tilt angle of flange= eval(Theta*180/@pi) [degrees]
###
### We define the default mesh-spacing,
### we define the borders of the computational volume.
###
define(Zmin, -4e-2)
define(Zmax, 4e-2)
-mesh
spacing= STPSZE
pxlow= -5e-2, pxhigh= 0
pylow= -4e-2, pyhigh= 4e-2
pzlow= Zmin, pzhigh= Zmax
#####
# Specify that the material index '3'
# describes a perfect conducting material.
-material
material= 3
type= electric
##
## fill the universe with metal
##
-brick
material= 3
xlow= -INF, xhigh= INF
ylow= -INF, yhigh= INF
zlow= -INF, zhigh= INF
doit
##
## Step 1: carve out a tilted circular box
##
-ggcylinder # a parallelogram gap with a tilt angle
material= 0
origin= (0,0,0)
xprimedirection= (1,0,0)
yprimedirection= (0, cos(Theta), sin(Theta) )
zprimedirection= ( 0, 0, 1 )
usezprimedirection= yes
range= ( -LL, LL )
clear
point= ( -RR, 0 )
arc, radius= RR,type= counterclockwise, size= small
point= ( RR, 0 )
arc, radius= RR,type= counterclockwise, size= smal
point= ( -RR, 0 )
show= later
doit
usezprimedirection= no # Switch back to the default, 'no'
##
## Step 2: Creating the interior footprint of elliptic beampipe(hollow)
##
-ggcylinder
material= 0
origin= (0, 0, 0)
xprimedirection= (1, 0, 0)
yprimedirection= (0, 1, 0)
range= (Zmin-2*STPSZE, Zmax+STPSZE)
xslope= 0, yslope= 0
clear # clear any old polygon-description of the footprint
# point= (x', y')
point= (0, -AxisB),
ellipse, center= (0,0),
point= (AxisA, 0),
ellipse,
point= (0, AxisB),
ellipse,
point= (-AxisA, 0),
ellipse,
point= (0, -AxisB),
# show= all
doit
############
-volumeplot
eyeposition= ( 1, -0.5, 0.5 )
scale= 3
doit
Example
The following decribes a cosine-taper. The cross-section is specified as a
ggcylinder, the cross section is circular.
The xscale and yscale are specified via tables.
# /usr/local/gd1/examples-from-the-manual/zxscale-example.gdf
define(R1, 1 )
define(R3, 3 )
define(DR, 0.1*R1)
-general
outfile= /tmp/UserName/bla
scratch= /tmp/UserName/scratch-
-mesh
spacing= R1 / 10
pxlow= -16, pxhigh= 5
pylow = -(R3+DR), pyhigh= R3+DR
pzlow = -(R3+DR), pzhigh= R3+DR
# Compile the program which creates the table.
system( $FC Zwo-Kosinus-Tabelle.f90 )
# Run the program which creates the table.
system( ./a.out > Two+Cosine-Table )
# The source code looks like:
# Pi= 4*ATAN(1.0)
# z0= 0
# zN= 10
# N= 100
# DO i= 1, N
# z= z0 + (i-1)*(zN-z0) / (N-1)
# WRITE (*,*) z, 2 - COS((z-z0) * Pi/(zN-z0))
# END DO
# END
#
##
## Create the tapered beampipe.
##
-ggcylinder
material= 3
origin= ( 0, 0, 0 )
xprime= ( 0, 0, 1 ) # the x'-direction.
yprime= ( 0, 1, 0 ) # The y'-direction.
# The z'-direction is then implicit the
# ( -1, 0, 0 ) direction.
# z' = x' cross y'
range= ( -3, 14 ) # the range of the z'-values.
# Filenames of the tables.
zxscale= Two+Cosine-Table
## zyscale= -none-
clear # clear any old polygon-description of the footprint
#
# This decribes a circular footprint.
#
# point= (x', y')
point= ( 0, -R1 )
arc, radius= R1, type= clockwise, size= small
point= ( 0, R1 )
arc
point= ( 0, -R1 )
# show= now
doit
-volumeplot, scale= 4, doit
gbor is a body of revolution with a cross section described
as a general polygon.
This cross section is swept along an axis in a general direction.
Moreover, only cells that fulfill some additional condition
will be filled.
############################################################################## # Flags: nomenu, noprompt, nomessage, # ############################################################################## # section -gbor # ############################################################################## # material = 1 # # whichcells = all, taboo= none # # show = off -- (off | all | later | now) # # name = gbor-000000000 # # inside = yes -- (yes|no) # # originprime = ( 0.0, 0.0, 0.0 ) # # zprimedirection= ( 0.0, 0.0, 1.0 ) # # rprimedirection= ( 1.0, 0.0, 0.0 ) # # range = ( 0.0, 360.0 ) # # xscaleprime = 1.0 -- Elliptic Coordinates. # # yscaleprime = 1.0 -- Elliptic Coordinates. # ############################################################################## ## syntax: # # point= (Zi, Ri) # # arc, radius= RADIUS, type= [clockwise | counterclockwise] # # size= [small | large ] # # deltaphi= 5 # # ellipse, center= (Z0, R0), size= [small | large ] # # deltaphi= 5 # ############################################################################## # doit, return, help, list, reset, clear # ##############################################################################
material= MAT:
whichcells
all, or a material-index.
whichcells=all, all volume inside the
gbor is assigned the material-index,
provided the former material is not taboo.
If whichcells is a material-index, only the parts of the
gbor that are currently filled with the given index
are assigned the new material-index.
taboo
none, or a material-index.
taboo=none, all volume inside the
gbor is assigned the material-index.
If taboo is a material-index, only the parts of the
gbor that are currently filled with another index
than the given index are assigned the new material-index.
originprime:
gbor.
zprimedirection:
gbor.
rprimedirection:
range:
xscaleprime, yscaleprime:
inside:
gbor
should be assigned material index MAT, or
whether the cells outside of it should be changed.
show:
show=off, no outline will be displayed.
show=later,
the outline of the gbor will be shown later, together
with outlines of other specified items.
If show=all is present,
the outlines of all other specified items bricks
gccylinders, ggcylinders and gbors
found in the inputstream so far where show
was not off will be displayed.
point= (XI, YI):
XI, YI are the coordinates of the i.th point in the polygon
that describes the polygon of the gbor.
There have to be minimum 3 points,
or 2 points and an arc
or 2 points and an ellipse.
arc:
radius= RADIUS:
RADIUS
size= [small | large] (optional):
type= [clockwise | counterclockwise]:
clear:
doit:
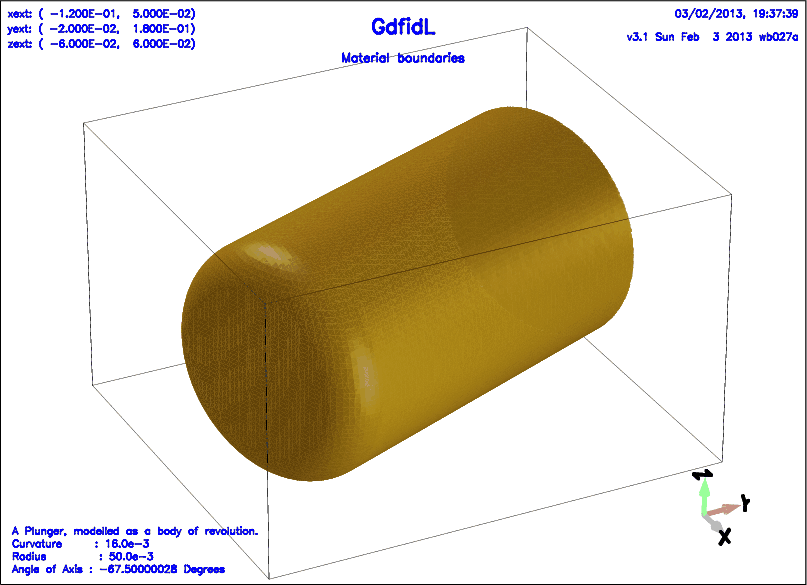
# /usr/local/gd1/examples-from-the-manual/plunger0.gdf
define(PlungerInnerRadius, 100e-3/2 )
define(PlungerCurvature, 16e-3 )
define(PlungerAngle, -67.5*@pi/180 )
-general
outfile= /tmp/UserName/example
scratch= /tmp/UserName/scratch-
text()= A Plunger, modelled as a body of revolution.
text()= Curvature : PlungerCurvature
text()= Radius : PlungerInnerRadius
text()= Angle of Axis : eval(PlungerAngle * 180 / @pi) Degrees
-mesh
spacing= 0.2e-2
pxlow= -0.12, pxhigh= 0.05
pylow= -0.02, pyhigh= 0.18
pzlow= -6e-2, pzhigh= 6e-2
-gbor
material= 4
originprime= ( 0, 0, 0 )
zprimedirection= ( cos(PlungerAngle), sin(PlungerAngle), 0 )
rprimedirection= ( 0, 0, 1 )
range= ( 0, 360 )
clear
# point= ( z, r )
point= ( 0, 0 )
point= ( 0, PlungerInnerRadius-PlungerCurvature )
arc, radius= PlungerCurvature, size= small, type= counterclockwise
point= ( -PlungerCurvature, PlungerInnerRadius )
point= ( -170e-3, PlungerInnerRadius )
point= ( -170e-3, 0 )
# show= now
doit
-volumeplot, eyeposition= ( 1, -0.5, 0.6 )
scale= 3
doit
# /usr/local/gd1/examples-from-the-manual/gbor-example1.gdf
define(LargeNumber, 10000) # some big number
define(MAXCELLS, 1e+5)
define(XLOW, 0) define(XHIGH, 5e-2)
define(YLOW, 0) define(YHIGH, 6e-2)
define(ZLOW, -1.1e-2) define(ZHIGH, 0)
define(STPSZE, ((XHIGH-XLOW)*(YHIGH-YLOW)*(ZHIGH-ZLOW)/MAXCELLS)**(1/3) )
-mesh
volume= ( XLOW, XHIGH, \
YLOW, YHIGH, \
ZLOW, ZHIGH )
spacing= STPSZE
-general
outfile= /tmp/UserName/example
scratch= /tmp/UserName/scratch
define(R, 1.0e-2)
text()= Intersection of two circular cylinders with radius R
text()= generated as general cylinders
text()= where 'taboo' is specified
text()= stpsze= STPSZE, maxcells= MAXCELLS
#
# Fill everything with metal
#
-brick
material= 1
volume= ( -LargeNumber, LargeNumber, \
-LargeNumber, LargeNumber, \
-LargeNumber, LargeNumber )
doit
#
# First step,
# fill cells above the diagonal with material 3
# these cells will not be filled by the first circular cylinder,
# since we will specify 'taboo= 3'
#
#
-ggcylinder
material= 3,
origin= ( 0, 0, 0 ), xprime= ( 1, 0, 0 ), yprime= ( 0, 1, 0 ),
range= ( ZLOW, ZHIGH ),
clear
point= ( XLOW, YLOW ), point= ( XLOW, YHIGH ),
point= ( XLOW+(YHIGH-YLOW), YLOW )
doit
#
# Second step:
# fill a circular cylinder in x-direction,
# but NOT cells with material index 3 (taboo=3)
#
-ggcylinder
material= 0, taboo= 3,
xprime= ( 0, 1, 0 ), yprime= ( 0, 0, 1 ), # so the axis will be in +x
origin= ( 0, 4e-2, 0 ), # shift of origin
range= ( XLOW, XHIGH ),
clear
point= ( -R, 0 ),
arc, radius= R, type= counterclockwise,
point= ( 0, -R ),
arc, radius= R,
point= ( R, 0 ),
doit
#
# Third step:
# fill a circular cylinder in y-direction,
# but ONLY cells with material index 3 (whichcells=3),
#
-ggcylinder
material= 0, whichcells= 3, taboo= none
xprime= ( 1, 0, 0 ), yprime= ( 0, 0, -1 ), # so the axis will be in +y
origin= ( 2.e-2, 0, 0 ), # shift of origin
range= ( YLOW, YHIGH ),
clear
point= ( -R, 0 ),
arc, radius= R, type= clockwise,
point= ( 0, R ),
arc, radius= R,
point= ( R, 0 ),
doit
-volumeplot
eyeposition= ( 1, 2, 1 )
scale= 3.5
doit
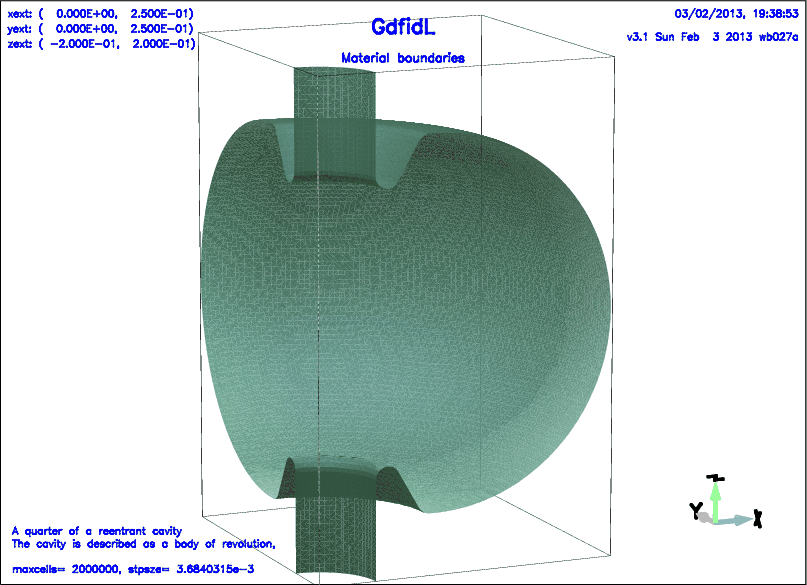
# /usr/local/gd1/examples-from-the-manual/gbor-example.gdf
define(MaxCells, 2e+6)
#
# define the geometry parameters
#
define(RBeamTube, 4.7625e-2)
define(RCurve, 1.0e-2)
define(ZGapNose, 10.9e-2)
define(RLarge, 25.0e-2) define(RSmall, 15.0e-2) define(RCenter, 10.0e-2)
define(INF, 10000) define(EL, 1) define(MAG, 2)
define(XLOW, -0.250) define(XHIGH, 0.250)
define(YLOW, -0.250) define(YHIGH, 0.250)
define(ZLOW, -0.200) define(ZHIGH, 0.200)
define(STPSZE, ((XHIGH-XLOW)*(YHIGH-YLOW)*(ZHIGH-ZLOW)/MaxCells)**(1/3) )
#
# gdfidl can evaluate sin(), cos(), atan() and X**Y
# definition of functions degsin() and degcos()
#
sdefine(degsin, [sin((@arg1)*@pi/180)])
sdefine(degcos, [cos((@arg1)*@pi/180)])
sdefine(degtan, [sin((@arg1)*@pi/180)/cos((@arg1)*@pi/180)])
-general
outfile= /tmp/UserName/example
scratch= /tmp/UserName/scratch-
text(3)= A quarter of a reentrant cavity
text()= The cavity is described as a body of revolution,
text()=
text()= maxcells= MaxCells, stpsze= STPSZE
-mesh
pxlow= 0*XLOW, pxhigh= 1*XHIGH
pylow= 0*YLOW, pyhigh= 1*YHIGH
pzlow= 1*ZLOW, pzhigh= 1*ZHIGH
cxlow= mag, cxhigh= mag
cylow= mag, cyhigh= mag
czlow= ele, czhigh= ele
spacing= STPSZE
-material
material= 3, type= electric
-brick
#
# we fill the universe with metal
#
material= 3
volume= ( -INF,INF, -INF,INF, -INF,INF )
doit
#
# carve out the cavity itself
#
-gbor
material= 0, range= ( 0, 360 )
origin= ( 0, 0, 0 )
show= later, # don't show now, but show later
clear # clear a possible previous polygon list
point= ( 0 , 0 ),
point= ( ZHIGH+2*STPSZE , 0 ),
point= ( ZHIGH+2*STPSZE , RBeamTube ),
point= ( ZGapNose+RCurve, RBeamTube ),
arc, radius= RCurve, size= small, type= clockwise,
deltaphi= 10
define(rdum, RBeamTube+(1+degcos(30))*RCurve)
define(zdum, ZGapNose +(1-degsin(30))*RCurve)
point= ( zdum, rdum )
define(deltaz, RSmall-zdum)
define(deltar, deltaz*degtan(30))
define(ffac, 0.85) ## adjust this for a smooth transition
define(zdum2, zdum+ffac*deltaz)
define(rdum2, rdum+ffac*deltar)
point= ( zdum2, rdum2 )
arc, radius= RCurve, size= small, type= counterclockwise,
deltaphi 10
point= ( RSmall, RCenter-0.8*RCurve ),
point= ( RSmall, RCenter ),
arc, radius= RSmall, size= small, type= counterclockwise,
delta= 3
point= ( -RSmall, RCenter ),
point= ( -RSmall, RCenter-0.8*RCurve ),
arc, radius= RCurve, size= small, type= counterclockwise,
deltaphi= 10
point= ( -zdum2, rdum2 )
point= ( -zdum, rdum )
arc, radius= RCurve, size= small, type= clockwise, delta 10
point= ( -(ZGapNose+RCurve), RBeamTube ),
point= ( ZLOW-2*STPSZE, RBeamTube ),
point= ( ZLOW-2*STPSZE, 0 )
list
doit
#
# enforce some meshplanes:
#
-mesh
zfixed( 2, -(ZGapNose+RCurve), -ZGapNose ) # at the noses
zfixed( 2, (ZGapNose+RCurve), ZGapNose ) # at the noses
zfixed( 2, -RSmall, RSmall) # at the z-borders of the cavity
-volumeplot
scale= 3
doit
Example
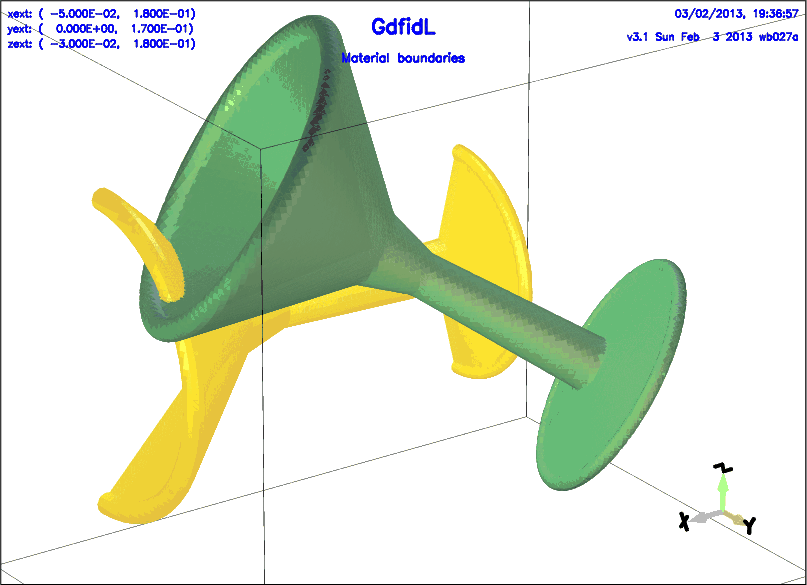
The following describes two bodies of revolution that look similiar to glasses.
The shape is only specified once, but two different glasses
are generated by varying 'zprime' and 'rprime'
# /usr/local/gd1/examples-from-the-manual/gbor-glasses.gdf
define(MAXCELLS,1e+6)
define(XLOW,-5e-2) define(XHIGH,18e-2)
define(YLOW,-0e-2) define(YHIGH,17e-2)
define(ZLOW,-3e-2) define(ZHIGH,18e-2)
define(STPSZE, ((XHIGH-XLOW)*(YHIGH-YLOW)*(ZHIGH-ZLOW)/MAXCELLS)**(1/3) )
-general
outfile= /tmp/UserName/glasses
scratch= /tmp/UserName/glasses-scratch-
-mesh
spacing= STPSZE
volume= ( XLOW, XHIGH, \
YLOW, YHIGH, \
ZLOW, ZHIGH )
# Fill the universe with vacuum
define(LargeNumber,10000)
-brick
material= 0
volume= ( -LargeNumber, LargeNumber, \
-LargeNumber, LargeNumber, \
-LargeNumber, LargeNumber )
doit
#
# The glasses:
#
-gbor
#
# Definition of the cross-section:
#
clear
point= ( 0.7e-2, 0 ),
point= ( 0, 4.0e-2 ),
arc, radius=0.25e-2, size= large, type= clockwise
point= ( 0.3e-2, 4.0e-2 ),
point= ( 1.0e-2, 1.0e-2 ),
point= ( 8.0e-2, 0.8e-2 ),
point= ( 10.0e-2, 1.4e-2 ),
point= ( 15.0e-2, 6.0e-2 ),
arc, radius= 0.4e-2, type= clockwise
point= ( 15.2e-2, 5.4e-2 ),
point= ( 10.0e-2, 0 )
#
# That cross-section is used twice,
# with different parameters for origin, zprime etc..
#
material= 1,
origin= ( -2e-2, 0, 4e-2 ),
zprimedir= ( 1, 0, 0 ),
rprimedir= ( 0, 1, 0 ),
range= ( -90, 90 ),
## show= now
doit # this 'doit' generates the first 'glass'
material= 3
origin= ( 0, 12e-2, 4e-2 ),
zprimedir= ( 1, -0.5, 0.7 ),
rprimedir= ( 0, 1, 0 ),
range= ( 0, 360 ),
## show= all
doit # this 'doit' generates the second 'glass'
-volumeplot
eyeposition= ( 0.7, 1, 0.5 )
scale= 4
doit
The so described body can be rotated, shrunk or expanded and shifted.
############################################################################## # Flags: nomenu, noprompt, nomessage, # ############################################################################## # section -stlfile # ############################################################################## # file= /usr/local/gd1/examples/woman.stl # # material = 1 # # whichcells= all, taboo= none, nboundaries= 1 # # originprime = ( 0.0, 0.0, 0.0 ) # # xprimedirection= ( 1.0, 0.0, 0.0 ) # # yprimedirection= ( 0.0, 1.0, 0.0 ) # # xscale = 1.0 # # yscale = 1.0 # # zscale = 1.0 # # debend = no -- debend description around uaxis # # uaxis= z -- [xyz] : direction of debend-axis # # radius= 0.0 # # v0 = 0.0 -- v0,w0: coordinates of the axis. # # w0 = 0.0 # # show = no -- ( yes | no ) # ############################################################################## # doit, return, help # ##############################################################################
file= NAME_OF_STLFILE:
material= MAT:
whichcells
all, or a material-index.
whichcells=all, all volume inside the
stl-body is assigned the material-index,
provided the former material is not taboo.
If whichcells is a material-index, only the parts of the
stl-body that are currently filled with the given index
are assigned the new material-index.
taboo
none, or a material-index.
taboo=none, all volume inside the
stl-body is assigned the material-index.
If taboo is a material-index, only the parts of the
stl-body that are currently filled with another index
than the given index are assigned the new material-index.
nboundaries= [1,2]
debend= [yes|no]
uaxis= [x|y|z]
debend=yes. The axis around which the body is bended.
radius= NUMBER
debend=yes. The bending radius.
v0= NUMBER, w0= NUMBER
debend=yes. The coordinates of the bending axis.
originprime= (X0, Y0, Z0): xprimedirection= (XXN, XYN, XZN): yprimedirection= (YXN, YYN, YZN): xscale= XS, yscale= YS, zscale= ZS:
The coordinates of a point P of the STL-set are now transformed via
P <= (A) * P
P_x <= P_x * xscale + X0
P_y <= P_y * yscale + Y0
P_z <= P_z * zscale + Z0
show= [yes|no] :
doit:
Example
# /usr/local/gd1/examples-from-the-manual/stl-example2.gdf
-mesh
spacing= 2.2
perfectmesh= yes
volume= ( 0,200, 0,130, -200,0 )
-stlfile
file= /usr/local/gd1/examples/wagner60kASTL.stl
xprime= ( 1, 0, 0 )
yprime= ( 0, 0, -1 )
material= 1, taboo= none
## show= yes,
doit
-volumeplot, scale= 2.5, eyepos= ( 1, 2, 0.5), doit
############################################################################## # Flags: nomenu, noprompt, nomessage, # ############################################################################## # section -geofunction # ############################################################################## # material = 1 # # whichcells= all, taboo= none # # fdescriptionsubroutine=-none- # # a1= 0.0, b1= 0.0 # # a2= 0.0, b2= 0.0 # # a3= 0.0, b3= 0.0 # # a4= 0.0, b4= 0.0 # # a5= 0.0, b5= 0.0 # # a6= 0.0, b6= 0.0 # # a7= 0.0, b7= 0.0 # # a8= 0.0, b8= 0.0 # # a9= 0.0, b9= 0.0 # ############################################################################## # doit, return, help # ##############################################################################
material:
whichcells:
taboo= [none|0..50]:
fdescriptionsubroutine= NAME-OF-A-SHARED-OBJECT:
SUBROUTINE fgeosub( Point, AArray, IsIn ) DOUBLE PRECISION, INTENT(IN), DIMENSION(1:3) :: Point DOUBLE PRECISION, INTENT(IN), DIMENSION(1:20) :: AArray INTEGER, INTENT(INOUT) :: InThe Point parameter are the cartesian components of the point, the AArray parameter are the numbers a1..a9, b1..b9, and the IsIn parameter is to be computed. It shall be zero, if the point is NOT in the body.
doit:
# /usr/local/gd1/examples-from-the-manual/geofunction-example.gdf
-general
outfile= /tmp/UserName/geofunction
scratch= /tmp/UserName/scratch
-mesh
perfectmesh= yes
spacing= 0.1e-3
pxlow= -3e-3, pxhigh= 3e-3
pylow= -3e-3, pyhigh= 3e-3
pzlow= 0, pzhigh= 5e-3
czlow= magnetic, czhigh= magnetic
pylow= 0, cylow= magnetic
-brick
material= 0, whichcells= all, taboo= none
volume= ( -INF,INF, -INF,INF, -INF,INF )
doit
if (1) then
# Fill Parts of the Universe which shall later be described
# by the 'geofunction'.
-brick
material= 10
xlow= -1e-3, xhigh= 1e-3
ylow= -INF, yhigh= INF
zlow= -INF, zhigh= INF
sloppy= yes
doit
xlow= -INF, xhigh= INF
ylow= -1e-3, yhigh= 1e-3
doit
sloppy= no
end if
system( ifort -O3 -fPIC -auto -c \
./PointInsideFunction.f90 )
system( gcc -fPIC -shared -Wl,-soname,PointInsideFunction.so \
-o /tmp/PointInsideFunction.so PointInsideFunction.o )
define(VAL, 1)
-geofunction
#
# If this Inputfile is to be used with PVM/MPI,
# each Task will try to load the Shared Object.
# We assume here, /tmp/ is accessible by each Task.
#
fdescriptionsubroutine= /tmp/PointInsideFunction.so
material= 3, whichcells= 10, taboo= none
## whichcells= all
a1= 1 # Key
a2= 1825283
a3= 692618
a4= 0.7 * 3.28e-3
a5= 0
a8= 1 # Key: Pot gt val
a9= VAL # Aequipotential-Value
doit
mat 4
a8= -1 # Key Pot lt val
a9= -VAL # Aequipotential-Value
doit
-brick
material= 0, whichcells= 10
volume= ( -INF,INF, -INF,INF, -INF,INF )
doit
-material
material= 3, type= electric
material= 4, type= electric
material= 10, type= electric
-volumeplot
scale= 4
eyepos ( -1, -2, 3 )
doit
-eigenvalues
estimation= 100e9
doit
The Sourcecode PointInsideFunction.f90:
SUBROUTINE fgeosub( Point, A, In )
DOUBLE PRECISION, INTENT(IN), DIMENSION(1:3) :: Point
DOUBLE PRECISION, INTENT(IN), DIMENSION(1:20) :: A
INTEGER, INTENT(INOUT) :: In
DOUBLE PRECISION :: Pi, Phi, P
DOUBLE PRECISION :: x, y, z, z0, z1, zz, az, rmz, oodL, a01, a10
!! DOUBLE PRECISION :: fAdaCOS
Pi= 4*ATAN(1.0d0)
IF (NINT(A(1)) == 1) THEN
x= Point(1)
y= Point(2)
z= Point(3)
P = A(4)+A(5)*z
Phi= A(2)*(x**2-y**2) &
+ A(3)*(x**2+y**2+(P/Pi)**2)*wbCos(2*Pi*z/P)
IF (A(8) > 0) THEN
IF (Phi > A(9)) THEN
In= 1
ELSE
In= 0
END IF
ELSE
IF (Phi < A(9)) THEN
In= 1
ELSE
In= 0
END IF
END IF
ELSE IF (NINT(A(1)) == 2) THEN
! a1,a2,a3,a4,a5,a6,a7,a8,a9 => A( 1: 9)
! b1,b2,b3,b4,b5,b6,b7,b8,b9 => A(11:19)
! Periode als Polynom dargestellt,
! Amplituden als Polynom dargestellt,
! Abschnittsweise
x= Point(1)
y= Point(2)
z= Point(3)
z0= A(2)
z1= A(3)
!c write (*,*) ' a(1:9):', a(1:9)
!c write (*,*) ' a(11:19):', a(11:19)
IF ((z < z0) .OR. (z > z1)) THEN
In= 0
RETURN
END IF
zz= z-z0
oodL= 1 / (z1-z0)
P = A(4) +zz*(A(5)-A(4))*oodL
az= A(6) +zz*(A(7)-A(6))*oodL
rmz= A(8)+zz*(A(9)-A(8))*oodL
a10= (rmz**2-1) &
/ (2*(rmz*az)**2+(rmz**2+1)*(P/Pi)**2)
a01= ((rmz**2+1)*az**2+2*(P/Pi)**2) &
/ (2*(rmz*az)**2+(rmz**2+1)*(P/Pi)**2) &
/ az**2
Phi= a01*(x**2-y**2) &
+ a10*(x**2+y**2+(P/Pi)**2)*wbCos(2*Pi*zz/P)
IF (A(11) > 0) THEN
IF (Phi > A(12)) THEN
In= 1
ELSE
In= 0
END IF
ELSE
IF (Phi < A(12)) THEN
In= 1
ELSE
In= 0
END IF
END IF
ELSE
In= 0
END IF
CONTAINS
DOUBLE PRECISION FUNCTION wbCos( x )
DOUBLE PRECISION, INTENT(IN) :: x
DOUBLE PRECISION :: xx
xx= ABS(x)
DO WHILE (xx > 2*Pi)
xx= xx - 2*Pi
END DO
IF (xx > Pi) THEN
Vorzeichen= -1
xx= xx - Pi
ELSE
Vorzeichen= 1
END IF
IF (xx > Pi/2) THEN
Vorzeichen= -Vorzeichen
xx= Pi - xx
END IF
wbCos= Vorzeichen* &
(1 - xx**2/2 + xx**4/(2*3*4) - xx**6 / (2.*3.*4.*5.*6.) )
!! + xx**8 / (2.*3.*4.*5.*6.*7.*8.) &
!! - xx**10 / (2.d0*3.d0*4.d0*5.d0*6.d0*7.d0*8.d0*9.d0*10.d0) )
END FUNCTION wbCos
END SUBROUTINE fgeosub
############################################################################## # Flags: nomenu, noprompt, nomessage, # ############################################################################## # Section: -transform # ############################################################################## # -translate # # -rotate # # # # Matrix: # # 1.00000000 , 0.00000000 , 0.00000000 , 0.00000000 # # 0.00000000 , 1.00000000 , 0.00000000 , 0.00000000 # # 0.00000000 , 0.00000000 , 1.00000000 , 0.00000000 # # 0.00000000 , 0.00000000 , 0.00000000 , 1.00000000 # ############################################################################## # reset, ?, return, help # ##############################################################################
-translate:
-rotate:
reset:
############################################################################## # Flags: nomenu, noprompt, nomessage, # ############################################################################## # Section: -translate # ############################################################################## # offset= ( 0.0, 0.0, 0.0 ) # # doit, ?, return, help # ##############################################################################
offset:
doit:
############################################################################## # Flags: nomenu, noprompt, nomessage, # ############################################################################## # Section: -rotate # ############################################################################## # axis= ( 0.0, 0.0, 1.0 ) # # angle= 90.0 # # Matrix: # # 1.00000000 , 0.00000000 , 0.00000000 , 0.00000000 # # 0.00000000 , 1.00000000 , 0.00000000 , 0.00000000 # # 0.00000000 , 0.00000000 , 1.00000000 , 0.00000000 # # 0.00000000 , 0.00000000 , 0.00000000 , 1.00000000 # ############################################################################## # doit, ?, return, help # ##############################################################################
axis:
angle:
doit:
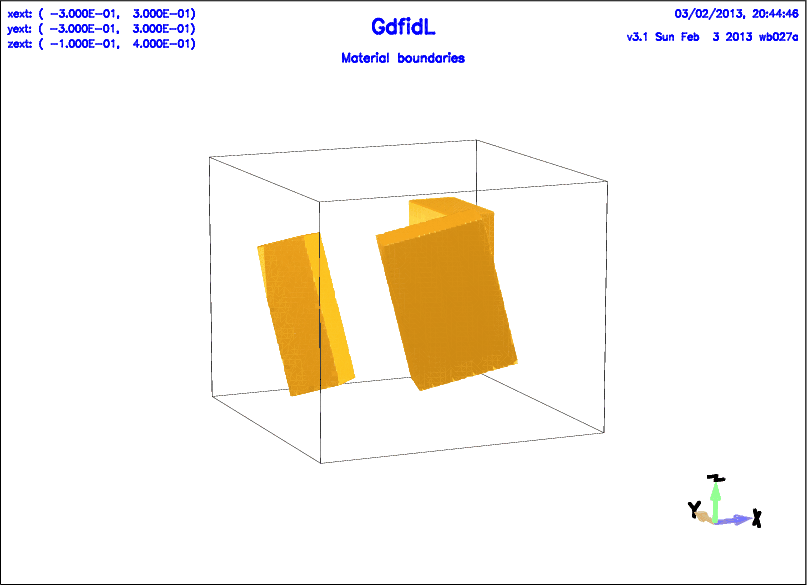
Example
-general
outfile= /tmp/UserName/bla
scratch= /tmp/UserName/scratch
-mesh
spacing= 0.01
pxlow= -0.3, pxhigh= 0.3
pylow= -0.3, pyhigh= 0.3
pzlow= -0.1, pzhigh= 0.4
-transform, reset
-brick
material= 1
volume= ( -BIG,BIG, -BIG,BIG, -BIG,BIG )
doit
#
# Translate all subsequent Items by (0.1, 0, 0).
#
-translate, offset= ( 1/10, 0, 0 ), doit
-brick
material= 0
name= brick1
xlow= 0, xhigh= 0.1
ylow= 0, yhigh= 0.2
zlow= 0, zhigh= 0.3
doit
#
# Additionally to the initial Translation,
# rotate by 90 Degrees around the (0,0,1)-Axis,
# then rotate 20 Degrees around the (0,1,0)-Axis,
#
-rotate,
axis= ( 0, 0, 1 ), angle= 90, doit
axis= ( 0, 1, 1 ), angle= 20, doit
#
# Next Brick, same Parameters as the first One,
# but additionally to the Translation now also rotated.
#
-brick, name= brick2, doit
#
# Next Rotation, same Parameters.
# Because the Rotations etc are all performed,
# subsequent Items are translated once and rotated four times.
#
-rotate,
axis= ( 0, 0, 1 ), angle= 90, doit
axis= ( 0, 1, 1 ), angle= 20, doit
# Next brick, same parameters as the previous one.
-brick, name= brick3, doit
-volumeplot, doit
############################################################################## # Flags: nomenu, noprompt, nomessage, # ############################################################################## # Section: -volumeplot # ############################################################################## # onlyplotfile = no -- Do not display Plot # # showpartition = no -- Show MPP-Partioning # # showlines = no # # text = yes # # scale = 1.80 # # plotopts = -geometry 690x560+10+10 # # eyeposition = ( -1.0, -2.30, 0.50 ) # # bbxlow = -1.0000e+30, bbylow = -1.0000e+30, bbzlow = -1.0000e+30 # # bbxhigh= 1.0000e+30, bbyhigh= 1.0000e+30, bbzhigh= 1.0000e+30 # # rotx = 0.0000 , roty = 0.0000 , rotz = 0.0000 # # # ############################################################################## # doit, ?, return, help # ##############################################################################
showpartition= [yes|no]:
showlines= [yes|no]:
text= [yes|no]:
text()= bla bla
in the section -general
should be plotted together with the material boundaries.
scale= SCALE:
plotopts= ANY STRING CONTAING OPTIONS FOR gd1.3dplot:
dataplot.ps.
eyeposition= (XEYE, YEYE, ZEYE):
bbxlow=,bbxhigh=,bbylow=bbyhigh=,bbzlow=,bbzhigh=:
rotx= PhiX, roty= PhiY, rotz= PhiZ:
PhiX,
then the result is rotated around the y-axis by an angle of PhiY,
and finally the result is rotated around the z-axis by an angle of PhiZ.
doit:
bbyhigh is specified such, that
only the material boundaries behind the plane y=0 are shown.
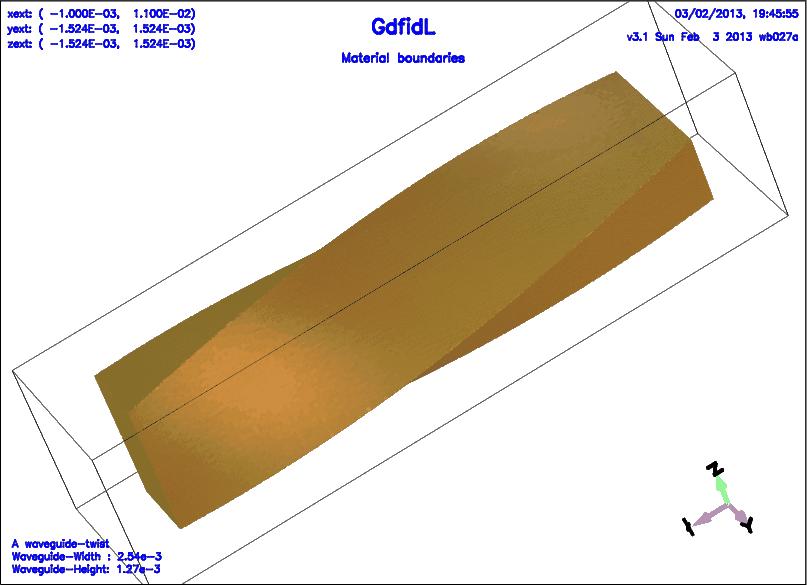
include(/usr/local/gd1/examples-from-the-manual/ggcylinder-twisted.gdf)
-volumeplot
eyeposition= ( 1, 2, 1.3 )
scale= 4.5
bbyhigh= 0
doit
Example
The following shows the material distribution of a waveguide twist again,
this time, the parameter roty is specified nonzero.
include(/usr/local/gd1/examples-from-the-manual/ggcylinder-twisted.gdf)
-volumeplot
eyeposition= ( 1, 2, 1.3 )
scale= 4.5
roty= 20
doit
A similar effect can be achieved with the section -volumeplot
with the aid of the parameters bb?low, bb?high.
############################################################################## # Flags: nomenu, noprompt, nomessage, # ############################################################################## # Section: -cutplot # ############################################################################## # onlyplotfile = no -- don't display plot # # draw = both -- both | approximated | input # # grid = yes -- yes | no # # normal = z -- plane normal of the cut-plane # # cutat = undefined -- the coordinate of the cut-plane # # # # plotopts = -geometry 690x560+10+10 # # eyeposition = ( -1.0, -2.30, 0.50 ) # ############################################################################## # doit, ?, return, help # ##############################################################################
draw= [both|approximated|input]:
grid= [yes|no]:
normal= [x|y|z], cutat= :
cutat: The coordinate value of the gridplane in direction
normal where the material filling shall be plotted.
plotopts= ANY STRING CONTAINING OPTIONS FOR gd1.3dplot:
dataplot.ps .
eyeposition= (XEYE, YEYE, ZEYE):
doit:
############################################################################## # Flags: nomenu, noprompt, nomessage, # ############################################################################## # section -eigenvalues # ############################################################################## # solutions = 15 # # estimation = undefined # # storeallmodes= no # # flowsave = 0.0 # # fhighsave = 1.0e+30 # # passes = 2 # # pfac2 = 1.0e-3 # # lossy = no -- Lossy or dispersive Computation # # flowsearch = undefined -- Edges of the search Region... # # fhighsearch= undefined -- ...when "lossy= yes" # # -ports -- ...when "lossy= yes" # # -linitialfields -- ...when "lossy= yes" # # # # # # compressed = no -- Minimal RAM, more CPU and IO Time # ############################################################################## # return, doit, help, ? # ##############################################################################
solutions= NSOL:
compressed= yes is specified, the memory requirement is NOT
proportional to the number of basisvectors, as then the basisvectors
are compressed and stored to disk and retrieved from disk.
The compressed basisvectors are written to files with names built from
the scratchbase parameter of section -general.
estimation= FUP:
NSOL.st mode.
storeallmodes= [yes | no]:
passes=1),
but does not store them.
If you do not believe that these static modes are really static,
you can enforce the storing to file with this option.
When the static solutions are in the file, you can view these
'solutions' with gd1.pp.
flowsave= FLOW, fhighsave= FHIGH:
-general, dice= yes, iodice= yes, which specifies that
the resultfields shall be stored on the local disks of the compute nodes.
BUT: -general, dice= yes, iodice= yes can only be used for the PVM version,
not the MPI version.
passes= NPASS:
NSOL basisvectors
that are mutually orthogonal and (hopefully) span
only the subspace also spanned by the eigenvectors
corresponding to the NSOL lowest eigenvalues.
passesNSOL.
Also, the accuracy of the fields becomes somewhat better.
pfac2= PFAC2:
pfac2,
that controls the relative cleaning of the basisvectors from eigenvectors
outside the range 0 to FUP, in which the NSOL resonant
fields are expected.
pfac2 leads to increased cpu-time, but better accuracy for
the lower modes. A good value is pfac2=1e-3.
lossy= [yes|no]:
lossy= yes, eigenvalues are searched in a geometry where the
lossy parameters and dispersive parameters of materials with
type= normal are taken into account.
This algorithm needs four times as much memory
as the lossfree algorithm.
flowsearch= F1, fhighsearch= F2:
lossy=yes:
The frequency range where the resonances of the lossy geometry are searched in.
-ports:
lossy= yes, absorbing boundary conditions are applied
at ports that are specified via -eigenvalues,-ports.
The section -eigenvalues,-ports is the same as the section
-fdtd,-ports.
-linitialfields:
lossy= yes, initial fields can be loaded as starting field.
The section -eigenvalues,-linitialfields is the same as the section
-fdtd,-linitialfields.
compressed:
compressed=yes, the basis vectors will be compressed and stored
to scratchfiles and re-read when needed.
This takes much less memory during the eigenvalue computation, but
increases substantial the Input-Output time.
doit:
doit starts the eigenvalue computation.
-eigenvalues solutions= 15 passes= 2 estimation= 1.3e9 doit
############################################################################## # Flags: nomenu, noprompt, nomessage, # ############################################################################## # Section: -fdtd # ############################################################################## # -ports -- Absorbing Boundary Conditions. # # -pexcitation -- Excited Port Modes. # # -lcharges -- One or more relativistic Line Charges. # # -windowwake -- Wakes in a moving Mesh Window. # # -clouds -- not yet finished. # # -linitialfields -- Loads initial Fields at t=0. # # -voltages -- Enforce Voltages between Points. # # -decaytime -- z-dependent Damping. # # -time -- Timestep etc. # # -storefieldsat -- When to store Fields. # # -fexport -- When to store Fields. # # -smonitor -- What Flux Quantities to store. # # -fmonitor -- What Field Quantities to store. # # -pmonitor -- What Power Quantities to store. # # # # hfdtd= no -- Use higher order Curl Operators. # # norder= 2 -- Order to use. 1..3 # ############################################################################## # doit, ?, return, help # ##############################################################################
-ports:
-fdtd/-ports where you specify the
location of "ports" and the properties of these ports.
-pexcitation:
-fdtd/-pexcitation, where you specify
the center frequency, the bandwidth and the amplitudes of selected
modes of selected ports.
-lcharges:
-fdtd/-lcharge, where you specify
the properties of relativistic line charges.
-windowwake:
-fdtd/-windowwake, where you specify
the properties of a moving mesh window to compute the wakepotential
of relativistic line charges.
-clouds:
-fdtd/-clouds, where you specify
the properties of a non-relativistic charge clouds.
As of today (October 2004), the implementation of this 'Particle in Cell'
computation is not yet fully finished.
Please contact Warner Bruns, if you need it badly.
-linitialfields:
-fdtd/-linitialfields,
where you specify what fields shall be the initialfields of a time
dependent computation.
-voltages:
-fdtd/-voltages, where you specify
excited voltages between arbitrary points.
-decaytime:
-time:
-fdtd/-time, where you specify
the minimum and maximum allowed simulation time.
-storefieldsat:
-fdtd/-storefieldsat.
You can specify multiple time-ranges where the electric and/ or magnetic
fields are stored to files.
-fexport:
-smonitor:
-fdtd/-smonitor, where you specify
what scalar quantities shall be monitored during the time domain
computation.
-fmonitor:
-fdtd/-fmonitor, where you specify
what field quantities shall be monitored during the time domain
computation.
-pmonitor:
-fdtd/-pmonitor, where you specify
what power quantities shall be monitored during the time domain
computation.
compressed:
compressed=yes, the FD-Coefficients will be used in a compressed
form. This takes much less memory during the time domain computation,
and normally also leads to a better performance. But it increases
the address space usage during the first few timesteps.
doit:
-eigenvalues,-ports.
A "port" is a part of the border of the computational volume that shall be treated as an infinitely long waveguide. In this section you specify the location of ports. You also specify the number of modes whose time amplitudes shall be written to file.
############################################################################## # Flags: nomenu, noprompt, nomessage, # ############################################################################## # Section: -ports # ############################################################################## # name = noname-001 # # plane = xlow # # modes = 1 # #(pxlow= -1.0e+30 , pxhigh= 1.0e+30 ) Ignored for plane=xlow # pylow= -1.0e+30 , pyhigh= 1.0e+30 # # pzlow= -1.0e+30 , pzhigh= 1.0e+30 # # epsmaximum= 2.0 # # muemaximum= 1.0 # # npml = 40 -- No of PML-Planes # # dampn = 50.0e-3 -- Damping factor in last Plane # # enforcemesh= yes -- Enforce translational invariant Mesh # ############################################################################## # doit, list, ?, return, help # ##############################################################################
name = ANY-STRING-WHICH-COULD-SERVE-AS-FILENAME:
plane= [xlow|xhigh|ylow|yhigh|zlow|zhigh]:
modes= NMODES:
No port amplitudes are monitored when performing a lossy eigenvalue computation.
pxlow, pxhigh, pylow, pyhigh, pzlow, pzhigh:
[x|y|z][low|high].
epsmaximum, muemaximum:
npml:
dampn:
doit:
list:
xlow1, xlow2.
Two ports are at the upper x-boundary of the computational volume,
the names are xhigh1, xhigh2.
The ports at the same boundary are distinguished by their pzlow, pzhigh
parameters.
-fdtd
-ports
name= xlow1, plane= xlow , pzlow= 0, modes= 10, doit
name= xhigh1, plane= xhigh, pzlow= 0, modes= 2, doit
name= xlow2, plane= xlow , pzhigh= 0, modes= 2, doit
name= xhigh2, plane= xhigh, pzhigh= 0, modes= 2, doit
############################################################################## # Flags: nomenu, noprompt, nomessage, # ############################################################################## # Section: -pexcitation # ############################################################################## # port = undefined # # mode = 1 # # amplitude = 1.0 # # phase = 0.0 # # frequency = undefined # # bandwidth = undefined # # risetime = 0.0 # # signalcommand= -none- # ############################################################################## # nextport, list, ?, return, help # ##############################################################################
port= NAME-OF-AN-ALREADY-DEFINED-PORT:
mode:
amplitude:
phase:
frequency:
bandwidth:
risetime:
list:
signalcommand= NAME_OF_COMMAND:
-none-, the excitation will be computed from
risetime, frequency, bandwidth and amplitude.
If specified as any other string, every 100 timesteps the specified
command is executed to compute the signal at the next 100 timesteps.
The command is executed as:
NAME_OF_COMMAND < ./gdfidl-actual-time-interval > ./gdfidl-actual-signal
The file ./gdfidl-actual-time-interval contains the following data:
iTime1, iTime2, TimeStep
iTime1: The number of the first timestep for which
the signal shall be defined.
iTime2: The number of the last timestep for which
the signal shall be defined.
TimeStep: The width of a timestep.
Beta, Alpha, DeltaZ.
Beta: The real part of the propagation constant of the
selected mode at the selected
frequency frequency.
Alpha: The imaginary part of the propagation constant.
DeltaZ: The width of the mesh at the selected port.
NAME_OF_COMMAND has to write to stdout
simply the values of the signal at the timesteps iTime1 to
iTime2, one line for each value.
nextport:
port, mode, amplitude and phase,
but the parameters frequency, bandwidth and risetime
are the same for all excited ports.
InputPort shall be excited.
Its amplitude shall be '1', the centerfrequency of the excited pulse shall be
1.2 GHz, and the bandwidth of the excited pulse shall be 0.7 GHz.
-fdtd,
-pexcitation
port= InputPort
mode= 1
amplitude= 1
frequency= 1.2e+9
bandwidth= 0.7e+9
InputPort1 shall be excited.
Its amplitude shall be '1', the frequency of the excited signal shall be
1.2 GHz, and the risetime until steady state of the excitation shall be
10 HF-periods.
In addition, the fundamental mode of the port with name
InputPort2 shall be excited. Amplitude shall be '1'.
-fdtd,
-pexcitation
port= InputPort1, mode= 1, amplitude= 1, phase= 0
frequency= 1.2e9, risetime= 10 / 1.2e9
nextport
port= InputPort2, mode= 1, amplitude= 1, phase= 0
Example
The following specifies that the fundamental mode of the port with name
InputPort shall be excited.
The portmode shall be computed for a frequency of 10 GHz,
and the signal of the excitation shall be defined by an external signal
command.
-fdtd,
-pexcitation
port= InputPort
mode= 1
amplitude= 1
frequency= 10 GHz
#
# compile the signalcommand
#
system($F90 signal-command.f -o signal-command)
signalcommand= ./signal-command
This is the sourcefile signal-command.f:
PROGRAM SignalCommand
IMPLICIT DOUBLE PRECISION (a-h,o-z)
Pi= 4*ATAN(1.0d0)
Frequency= 10e9
TRise= 10/Frequency
TDecay= 20/Frequency
THold= 50/Frequency
READ (*,*) iTime1, iTime2, TimeStep
READ (*,*) Beta, Alpha, DeltaZ
DO iTime= iTime1, iTime2, 1
ActualTime= iTime*TimeStep
IF (ActualTime .LE. TRise) THEN
Phi= ActualTime * Pi / TRise
Factor= (1-COS(Phi))/2
ELSE IF (ActualTime .LE. TRise+THold) THEN
Factor= 1
ELSE IF (ActualTime .LE. TRise+THold+TDecay) THEN
Phi= (ActualTime - (TRise+THold)) * Pi / TDecay
Factor= (1+COS(Phi))/2
ELSE
Factor= 0
ENDIF
Factor= Factor * TimeStep / DeltaZ
WRITE (*,*) Factor*SIN(2*Pi*Frequency*ActualTime)
ENDDO
END PROGRAM SignalCommand
The standard usage of this section is to compute wakepotentials. When a nonzero charge is specified, and a time domain computation is performed, the wakepotentials are computed as the potential that is seen by witness particles that are traveling in positive z-direction.
############################################################################## # Flags: nomenu, noprompt, nomessage, # ############################################################################## # Section: -lcharge # ############################################################################## # ----- Parameters of a line charge: ------ # # charge = 0.0 -- [As] # # shape = gaussian -- gaussian | triangular | table # # tablefile= -none- # xtable = 1.0 -- [m] # # sigma = undefined -- [m] # # isigma = 6 -- number of sigmas to use. # # xposition = 0.0 -- [m] # # yposition = 0.0 -- [m] # # direction = +z -- +z, -z # # soffset = 0.0 -- [m] # # ----- other parameters: ------ # # shigh = auto -- [m] # # showdata = no -- ( yes | no ) # # napoly = yes -- ( yes | no ) # # naccuracy= 10.0e-6 -- wanted acc for poisson-eq in Napoly alg. # # ignoretem= yes -- should be "yes" for Wx, Wy # ############################################################################## # ?, nextcharge, return, help # ##############################################################################
charge:
shape:
gaussian, triangular and table.
tablefile:
sigma is then ignored.
xtable:
sigma:
isigma:
-windowwake it might be useful to specify
isigma=3. Then, the length of the computational window extends from
-3*isigma before the center of the charge up to shigh behind the center of the charge.
xposition, yposition:
direction:
soffset:
soffset has the effect that the charge travels
later through a given plane.
ds:
auto" or a positive real.
If "ds= auto", a value of "spacing/2" is used
("spacing" is defined in -mesh).
When napoly= yes, the value for ds is fixed to c*dt,
where c is the velocity of light and dt is the used timestep.
This parameter is not very useful. Please ignore it.
shigh:
auto" or a positive real.
If "shigh= auto", a value of "20 * sigma" is taken.
showdata= [yes|no]:
napoly= [yes|no]:
napoly= yes, near the z-borders of the computational volume,
an integration of the wakefield similiar to the Napoly-integration is
performed. This option should be set to napoly= yes for scraper-like
structures.
One can keep the option set even
if it is no needed, as GdfidL checks whether such a integration is useful.
If it is not, it is not done.
naccuracy= SMALL-NUMBER:
ignoretem= [yes|no]:
ignoretem=yes, the term proportional to the primary field
of the exciting charge is ignored.
nextcharge:
-lcharge
xpos= 0
ypos= 0
charge= 1e-12
sigma= 1e-3
shigh= 100e-3
Example
An example for a table file describing a charge shape is given below.
# -lcharge # shape= table, tablefile= Bunch1.txt # xtable= 1e-6, charge= 1e-12 # # Everything behind a '#' is ignored. # Lines in the tablefile may be empty, or they may # contain two (or more) numbers. # The first number is a position, the next number is # a relative charge. What follows behind these two numbers is ignored. # # bunch1 (linac entrance) # s[microns] No. of macro-particles # ________________________________________________ 0, 0 1 -1.32067 2 -1.10711 3 -0.894051 4 -0.6815 5 -0.469454 6 -0.25791 7 -0.046869 8 0.16367 9 0.373709 10 0.583247
############################################################################## # Flags: nomenu, noprompt, nomessage, # ############################################################################## # Section: -windowwake # ############################################################################## # strangsplitting= yes # # fdtdorder= 31 -- What Order of FDTD Algorithm to take [1,31] # # periodic = no -- Assume periodic Geometry. # # nperiods= 100 -- Maximum Number of Periods to consider. # # modstore= 10 -- Store Wakepotentials after simulating # # -- Multiples of modstore*(pzhigh-pzlow). # # __xpartition= no -- PVM/MPI: Enforce Partitioning in x only. # ############################################################################## # doit, ?, return, help # ##############################################################################
strangsplitting:
yes, no".
strangsplitting=yes, a scheme with splitted updates for the
z-transverse components and z-longitudinal components is used.
This scheme has negligible dispersion error for TEM-waves propagating
in z-direction. The dispersion error for other kind of waves is comparable
to a first order FDTD algorithm
fdtdorder:
strangsplitting=no, and fdtdorder=1, the normal FDTD
algorithm is used to advance the electromagnetic fields.
If strangsplitting=no, and fdtdorder=31, a modified FDTD
algorithm is used to advance the electromagnetic fields.
That scheme has zero dispersion order for waves going in x- or y- or z-direction
and less dispersion error than strang-splitting for all kind of waves.
periodic:
yes, no".
periodic= no, the length of the geometry as specified
via
-mesh, pzlow= ZLOW, pzhigh= ZHIGH is used for computing the
wakepotential.
periodic= yes, the length of the geometry as specified
via
-mesh, pzlow= ZLOW, pzhigh= ZHIGH is assumed to be one period
of a finite periodic structure with number of periods given by
nperiods= NP. After computing the fields in a multiple of
modstore= MP periods, the wakepotentials so far are recorded
to the database.
nperiods= NP:
periodic=yes.
modstore= MP:
-lcharge. The moving computational window has a length of
isigma*sigma + shigh.
The grid-spacing in z is the minimum of all the x-spacings and y-spacings
and, if the specified "zspacing" in section "-mesh" is smaller, then that
zspacing is used.
Napoly-like integration is performed, and cannot be switched off.
The memory requirement is proportional to the length of the moving computational window, i.e. proportional to isigma*sigma + shigh. One should not specify a large value for shigh, in particular not longer than the structure length, otherwise a conventional wakepotential computation is more economic.
The CPU requirement is proportional to the length of the moving window times the length over which the moving window must travel. That length is the z-extension of the structure plus the length of the window. The CPU requirement is proportional to (isigma*sigma + shigh) * (pzhigh-pzlow + isigma*sigma + shigh). Any specified port is ignored. The x- and y-extension of the computational volume must be specified large enough that reflections from the x- and y-borders cannot change the wakepotentials. E.g. waveguides going in x- or y-direction should be modeled with a length of "shigh".
Lossy dielectrics are ignored.
Only fields specified via fexport will be exported.
The only other result is the wakepotential.
Example
The following specifies that we want to compute the wakepotential
of a linecharge travelling with the speed of light along the axis (x,y)=(0,0).
The total charge of the line-charge shall be 1 pC, and its gaussian width
sigma shall be 1 mm.
We are interested in s-values up to 20 mm.
-lcharge
xpos= 0, ypos= 0
charge= 1e-12
sigma= 1e-3
shigh= 20e-3
-windowwake
doit
If a 'Particle in Cell' computation is badly needed, contact Warner Bruns Field Computations. It might be that by the date that you read this, what you need is already available.
############################################################################## # Flags: nomenu, noprompt, nomessage, # ############################################################################## # Section: -clouds # ############################################################################## # box = no # # fraction = 0.950 # # bverbose = no # # ejectioncommand= -none- # # ejectionsubroutine= -none- # # # # # ############################################################################## # ?, return, help # ##############################################################################
The initial field is the sum of the specified fields.
############################################################################## # Flags: nomenu, noprompt, nomessage, # ############################################################################## # Section: -linitialfields # ############################################################################## # infile = ./previous_outfile # # symbol = e_1 # # quantity = e # # solution = 1 # # factor = 1.0 # # torealpart= yes # # static = no # # brz = no # # abstatic = 0.0 # # nbstatic = ( 0.0, 0.0, 1.0 ) # ############################################################################## # doit, ?, return, end, help # ##############################################################################
infile= NAME_OF_A_PREVIOUSLY_USED_OUTFILE:
infile via the section
-lgeometry.
symbol= QUAN_ISOL:
quantity= QUAN:
solution= ISOL:
factor:
torealpart:
static:
static= yes, the loaded field will only be used to accelerate
or deflect free moving charges in the PIC-algorithm.
If static= no, the loaded field will be used as part of the
initial condition for the electromagnetic field.
brz:
abstatic:
nbstatic:
doit:
#
# As we need the filename in two places better be the same,
# We define the files name as a variable.
#
sdefine( INFILE, /tmp/UserName/resonant-computation )
-lgeometry
infile= INFILE
-linitialfields
infile= INFILE
symbol= e_1, factor= 1, doit
symbol= h_1, factor= -1, doit
-voltages section.
############################################################################## # Flags: nomenu, noprompt, nomessage, # ############################################################################## # Section: -voltages # ############################################################################## # startpoint = ( 0.0, 0.0, 0.0 ) # # endpoint = ( 0.0, 0.0, 0.0 ) # # resistance = 0.10 -- [Ohm] # # inductance = 0.0 -- [Henry] # # amplitude = undefined -- [V] # # phase = 0.0 -- [Degs] # # frequency = undefined -- [1/s] # # bandwidth = undefined -- [1/s] # # risetime = undefined -- [s] # # logcurrent = yes # # name = noname-001 # ############################################################################## # doit, ?, list, return, help # ##############################################################################
startpoint, endpoint:
resistance:
inductance:
amplitude, phase, frequency, bandwidth:
risetime:
risetime, the voltage is switched on according to the formula:
 |
(1.6) |
logcurrent:
name:
doit:
If voltage sources are specified, the electric field strength between the startpoint
and endpoint is measured while the time domain computation is running.
The difference between the specified voltage and the so measured voltage
is multplied by the resistance of the voltage source.
That value is used as current which is enforced to flow between the starting
point and endpoint. The current is low pass filtered with a time constant given by ![]() .
.
The values of the wanted voltage, the measured voltage and the current can be inspected in the postprocessor.
No example yet.
############################################################################## # Flags: nomenu, noprompt, nomessage, # ############################################################################## # section -decaytime # ############################################################################## ## syntax: # # point= (Z_i, Tau_i) # ############################################################################## # clear, show, doit, ?, return, help # ##############################################################################
point:
clear:
show:
Example
-fdtd
define(FREQ, 12e9)
-decaytime
clear
point= ( -INF, 6000 / (@pi*FREQ) )
point= ( Zmin, 6880 / (@pi*FREQ) )
point= ( Zmax, 6580 / (@pi*FREQ) )
point= ( INF, 6000 / (@pi*FREQ) )
# show
doit
############################################################################## # Flags: nomenu, noprompt, nomessage, # ############################################################################## # section -time # ############################################################################## # firstsaved = undefined # # lastsaved = undefined # # distancesaved = undefined # # tminimum = undefined # # tmaximum = undefined # # decaytime = 1.0e+30 # ############################################################################## # amptresh = 3.0e-3 # # dtsafety = 0.90 # # ndt = auto # # ___stopafter= 1.0e+30 -- Force Stop after that Time. # # ___evmax = undefined -- Dangerous. # # ___dt = undefined -- Dangerous. # ############################################################################## # return, help, ? # ##############################################################################
firstsaved:
lastsaved:
distancesaved:
tminimum= TMIN, tmaximum= TMAX:
TMIN and a maximum one of TMAX.
TMIN no power is flowing through any port,
the simulation is stopped.
TMIN are power flowing through all ports is
stationary, the simulation is stopped.
TMAX, the simulation will be stopped.
decaytime= OOALPHA:
OOALPHA:
The reason for this parameter is as follows:
single.gd1 calculates mostly in single precision,
with a relative accuray of its internal values of ![]() for most
machines.
With this machine accuracy, it is not possible to calculate
the decay of high Q-structures.
If you would fill your volume not with vacuum but with a dielectric with
a very low conductivity
for most
machines.
With this machine accuracy, it is not possible to calculate
the decay of high Q-structures.
If you would fill your volume not with vacuum but with a dielectric with
a very low conductivity ![]() ,
the program would attempt to scale in every timestep the electric field
with a factor of
,
the program would attempt to scale in every timestep the electric field
with a factor of
 . For low values of
. For low values of ![]() this factor is so near to one that
it cannot be represented in single precision.
this factor is so near to one that
it cannot be represented in single precision.
With the specification of decaytime,
the fields (both E and B) in the entire volume are scaled
by such a factor, that after a simulation time of OOALPHA
the fields have decayed by a factor of 1/e.
If you have to calculate the scattering parameters of
a resonator with a known (not too low) internal Q-value,
e.g. greater than 1000, you can specify artificial losses with this
keyword and gd1 or single.gd1 together with gd1.pp
will calculate the proper scattering parameters.
amptresh= TRESH:
dtsafety:
dtsafety.
The used timestep is
ndt:
___stopafter:
___evmax:
___evmax.
NOT USEFUL. This is avaiable to test that the results of parallel and serial computations are bitwise identical.
___dt:
define(FREQ, 1e9) tmin= 100/FREQ, tmax= 1000/FREQ firstsaved= 10/FREQ, lastsaved= 20/FREQ distance= 1/2/FREQ
-fmonitor or -smonitor.
############################################################################## # Flags: nomenu, noprompt, nomessage, # ############################################################################## # Section: -storefieldsat # ############################################################################## # name = noname-0001 # # whattosave= both -- e-fields, h-fields, both, clouds # # firstsaved = undefined # # lastsaved = undefined # # distancesaved= undefined # ############################################################################## # doit, list, ?, return, help # ##############################################################################
name:
"name= a", the saved e-fields are
accessible later in the post-processor under the name "a_e_1" for the first
stored field. The first stored h-field of the set is accessible as
"a_h_1".
whattosave:
e-fields, h-fields, both, clouds.
The clouds value is for PIC-computations, it specifies that only the
data of free moving charges shall be stored at the selected times.
If you have a PIC-computation running, and you have selected
one of e-fields, h-fields, both, the data of free moving charges
are stored as well.
firstsaved:
lastsaved:
distancesaved:
doit:
list:
define(FREQ, 1e9)
-fdtd
-storefieldsat
name= a, what= both
firstsaved= 10/FREQ
lastsaved= 12/FREQ
distance= 0.1/FREQ
doit
name= b, what= e-fields
firstsaved= 20/FREQ
lastsaved= 30/FREQ
distance= 0.25/FREQ
doit
define(FREQ, 1e9)
-fdtd
-storefieldsat
do ii= 100, 1000, 100
#
# This 'name= a-ii' gets expanded to eg. 'name= a-100'
#
name= a-ii, what= e-fields
firstsaved= ii /FREQ
lastsaved= (ii+1+1/8)/FREQ
distance= 1/8/FREQ
doit
enddo
############################################################################## # Flags: nomenu, noprompt, nomessage, # ############################################################################## # Section: -fexport # ############################################################################## # outfile= noname-0001 # whattosave= e -- e-fields, h-fields, honmat # # firstsaved = undefined # # lastsaved = undefined # # distancesaved= undefined # # bbxlow = -1.0000e+30, bbylow = -1.0000e+30, bbzlow = -1.0000e+30 # # bbxhigh = 1.0000e+30, bbyhigh= 1.0000e+30, bbzhigh= 1.0000e+30 # ############################################################################## # doit, ?, return, end, help # ##############################################################################
outfile:
whattosave:
whattosave=honmat, the values of H on electric conducting
surfaces are written in a complicated special format, which can only be
read by gd1.pp's section -3dmanygifs.
firstsaved, lastsaved, distancesaved:
bbxlow=,bbxhigh=,bbylow=bbyhigh=,bbzlow=,bbzhigh=:
doit:
define(STPF, 0.5)
define(NP, 4)
define(RADIUS, 1+1)
define(GAP, 0.5)
define(PERIODE, 0.6)
define(BEAMR, RADIUS/2)
define(STPSZE, SIGMA/10/STPF)
define(DRADIUS, 2*STPSZE )
define( ZLOW, -(NP*PERIODE+BEAMR)/2 )
define( ZHIGH, (NP*PERIODE+BEAMR)/2 )
-general
outfile= /tmp/UserName/resultfile
scratch= /tmp/UserName/scratch
-mesh
pxlow= -(RADIUS+DRADIUS), pxhigh= (RADIUS+DRADIUS)
pylow= -(RADIUS+DRADIUS), pyhigh= (RADIUS+DRADIUS)
pzlow= ZLOW, pzhigh= ZHIGH
define(PZLOW, ZLOW)
define(PZHIGH, ZHIGH)
pxlow= 0, cxlow= mag
pylow= 0, cylow= mag
spacing= STPSZE
############
############
-brick, material 1, volume (-INF, INF, -INF, INF, -INF, INF), doit
do ip= -(NP-1)/2, (NP-1)/2, 1
-gccylinder
material= 0, radius= RADIUS, length= GAP
origin= (0,0,ip*PERIODE-GAP/2)
direction= (0,0,1)
show= later
doit
enddo
-gccylinder
material= 0, radius= BEAMR, length= INF
origin= ( 0, 0, -INF/2 ), direction= ( 0, 0, 1 )
# show= all
doit
#####
-volumeplot, scale 1.8, plotopts -geometry 600x550+10+10, doit
####
-lcharge
sigma= 0.1, charge= 1e-12, xposition= 0, yposition= 0
#
# The following large value for shigh is chosen for getting a movie of the
# full structure.
#
shigh= ZHIGH-ZLOW
define(NDT, 0.5 * SIGMA / @clight)
-fexport
outfile= /tmp/UserName/H-onmat-
what= honmat
firstsaved= 1e-20, lastsaved= 1e20
distancesaved= NDT
bbxlow= 0, bbxhigh= INF
bbylow= 0, bbyhigh= INF
bbzlow= PZLOW+2*STPSZE, bbzhigh= PZHIGH-2*STPSZE
doit
-windowwake,
doit
end
#####################
The following is input for gd1.pp to read the files and create many gifs from them. From the gifs, a mpeg file is created and displayed.
# Input for gd1.pp: -3dmanygifs 1stinfile= /tmp/UserName/H-onmat--000000001.gz outfiles= /tmp/UserName/absh- ## uptonfiles= 10 # what= abs what= logabs uptonfiles= 1e9 xrot= -30, yrot= 40 doit system( mpeg_encode ./gdfidl.3dmanygifs-mpeg_encode-params ) system( mpeg_play -dither color fexported.mpg )
############################################################################## # Flags: nomenu, noprompt, nomessage, # ############################################################################## # Section: -smonitor # ############################################################################## # name = noname-001 # # whattosave= convectioncurrent # # -- convectioncurrent, displacementcurrent # # -- magneticflux # # -- poyntingflux, ppclouds, pnclouds # # normal= z, cutat= undefined # # xlow= -1.0e+30 , xhigh= 1.0e+30 # # ylow= -1.0e+30 , yhigh= 1.0e+30 # # zlow= -1.0e+30 , zhigh= 1.0e+30 # ############################################################################## # doit, list, ?, return, help # ##############################################################################
name:
whattosave:
normal:
cutat:
bbxlow=,bbxhigh=,bbylow=bbyhigh=,bbzlow=,bbzhigh=:
doit:
############################################################################## # Flags: nomenu, noprompt, nomessage, # ############################################################################## # Section: -fmonitor # ############################################################################## # name = noname-001 # # whattosave= ecomponents # # -- ecomponents, hcomponents # # position= ( 0.0e-93, 0.0e-93, 0.0e-93 ) # ############################################################################## # doit, list, ?, return, help # ##############################################################################
name:
whattosave:
position:
doit:
############################################################################## # Flags: nomenu, noprompt, nomessage, # ############################################################################## # Section: -pmonitor # ############################################################################## # name = noname-001 # # whattosave= pdielectrics # # -- ( pdielectrics | energy ) # # xlow= -1.0e+30 , xhigh= 1.0e+30 # # ylow= -1.0e+30 , yhigh= 1.0e+30 # # zlow= -1.0e+30 , zhigh= 1.0e+30 # # stride= 10 # ############################################################################## # doit, list, ?, return, help # ##############################################################################
name:
whattosave:
xlow=, xhigh=, ylow=, yhigh=, zlow=, zhigh= :
doit:
-sparameter)
computes scattering parameters from the amplitudes of port-modes
that were computed and stored by
gd1.
-wakes.
-totouchstone.
Before you can do anything useful, you have to specify from what database
gd1.pp shall take the fields from. Therefore the first thing you do is:
enter the section -general.
############################################################################## # Flags: nomenu, noprompt, nomessage, # ############################################################################## # Section: -base # ############################################################################## # -general -- Define Database # # -3darrowplot -- Field Plots, Material Plot # # -lineplot -- Lineplot of Field Components, # # -glineplot -- Lineplot of Field Comp along gline, # # -2dplot -- Aquivalue Plot of a Component on a Plane. # # -energy -- Compute stored Energy # # -lintegral -- Compute Voltages # # -wlosses -- Compute Wall Losses # # -dlosses -- Compute dielectric Losses # # -flux -- Compute Fluxes through rectangular Areas # # -clouds -- Properties of free moving Clouds # # -sparameters -- Analyses Time History of Port Mode Amplitudes # # -material -- Specify Conductivities for Loss Computations # # -wakes -- Wakepotentials, Impedances # # ***** Miscellanea ****** # # -totouchstone -- Convert fri-data to touchstone Form # # -combine -- Combines Scattering Parameters # # -pcombine -- Combines E & H to Poynting Field. # # -smonitor -- Display and fft smonitor Data # # -fmonitor -- Display and fft fmonitor Data # # -pmonitor -- Display pmonitor Data # # -fexport -- Export 3D Field Data # # -2dmanygifs -- # # -3dmanygifs -- # # -debug -- Specify Debug Levels # # # ############################################################################## # ?, help, end, ls # ##############################################################################
-general:
-3darrowplot:
-lineplot:
-glineplot:
-2dplot:
-energy:
-lintegral:
-wlosses:
-dlosses:
-flux:
-clouds:
-sparameters:
-material:
-wakes:
-totouchstone:
-combine:
-smonitor:
-fmonitor:
-pmonitor:
-fexport:
-2dmanygifs, -3dmanygifs:
-fexport.
-debug:
############################################################################## # Flags: nomenu, noprompt, nomessage, # ############################################################################## # Section: -general # ############################################################################## # infile = ./results # # scratchbase= ./scratch. # # text( 1)= # ############################################################################## # plotopts = -geometry 690x560+10+10 # # showtext = yes -- (yes | no) # # onlyplotfiles= no -- (yes | no) # # nrofthreads= 4 -- SMP: Nr of Threads. # ############################################################################## # ?, return, end, help, ls # ##############################################################################
infile:
scratchbase:
TMPDIR set,
gd1.pp will take as default value set scratchbase to the string
$TMPDIR/gdfidl-scratch-pid=XXXXX-
Here $TMPDIR is the value of the environment variable TMPDIR,
and XXXXX
is the number of the process-id of gd1.pp.
text()= ANY STRING, NOT NECESSARILY QUOTED
text(NUMBER)= ANY STRING, NOT NECESSARILY QUOTED ANY STRING, NOT NECESSARILY QUOTED:
NUMBER:
NUMBER, the string following text()=
is placed in the next free line.
In the case with NUMBER, it is guaranteed, that the string
is placed in the NUMBER.st line.
You can specify up to 20 annotation strings,
the maximum length of each annotation string is 80 characters.
When a new database is specified, the values of text are
overwritten by the values that are found in the database.
plotopts= ANY STRING CONTAING OPTIONS FOR THE PLOT-PROGRAMS:
showtext= [yes|no]:
onlyplotfiles= [yes|no]:
-general
infile= @last
scratchbase= /tmp/garbage/delete-me-
plotopts= -geometry 1024x768
############################################################################## # Flags: nomenu, noprompt, nomessage, # ############################################################################## # Section: -3darrowplot # ############################################################################## # symbol = e_1 # # quantity = e # # solution = 1 # # component = all -- (x|y|z|all) # # phase = 45.0 -- Only for Portmodes # # arrows = 10000 # # lenarrows = 2.0 # # maxlenarrows= 2.0 # # fcolour = 4 # # materials = yes -- (yes | no) # # dielectrics = yes -- (yes | no) # # fonmaterials= no -- (yes | no) # # logfonmat= no -- (yes | no) # # fmaxonmat= auto # # scale = 2.80 # # fscale = auto # # nlscale = no, nlexp= 0.30 # # eyeposition = ( -1.0, -2.30, 0.50 ) # # bbxlow = -1.0000e+30, bbylow = -1.0000e+30, bbzlow = -1.0000e+30 # # bbxhigh= 1.0000e+30, bbyhigh= 1.0000e+30, bbzhigh= 1.0000e+30 # # rotx = 0.0000, roty = 0.0000, rotz = 0.0000 # # clouds = yes -- Show Clouds # ############################################################################## # plotopts = -geometry 690x560+10+10 # # showtext = yes -- (yes | no) # # showlines = no -- (yes | no) # # onlyplotfiles= no -- (yes | no) # ############################################################################## # @absfmax: undefined # ############################################################################## # doit, ?, return, end, help, ls # ##############################################################################
symbol= QUAN_ISOL:
quantity= QUAN:
solution= ISOL:
component:
x,y,z,all.
Only the selected component will be plotted.
phase:
arrows:
lenarrows:
lenarrows= 1, the arrows are scaled such, that for an homogeneous
field the end of one arrow is just behind the end of the next arrow.
maxlenarrows:
fcolour= :
materials= :
yes, the material boundaries are plotted together with the
fields.
dielectrics= :
dielectrics= no, material boundaries between dielectrics
will not be shown.
fonmaterials= :
logfonmat= :
maxfonmat= :
scale= SCALE:
fscale:
auto is specified, the vectorfield is scaled by
this value.
No further scaling takes place.
This is useful for generating movies, where every frame of the movie should
be scaled by the same factor.
nlscale, nlexp:
nlscale= yes, the size of field arrows are proportional to
the amplitude of the field to the power of nlexp.
eyeposition= (XEYE, YEYE, ZEYE):
bbxlow=,bbxhigh=,bbylow=bbyhigh=,bbzlow=,bbzhigh=:
rotx= PhiX, roty= PhiY, rotz= PhiZ:
PhiX,
then the result is rotated around the y-axis by an angle of PhiY,
and finally the result is rotated around the z-axis by an angle of PhiZ.
clouds= :
plotopts= ANY STRING CONTAING OPTIONS FOR THE PLOT-PROGRAMS:
showtext= [yes|no]:
showlines= :
yes, thin lines are plotted around the material patches,
indicating the used discretisation.
onlyplotfiles= [yes|no]:
doit:
symbol= e_1 doit
symbol= ere_4 fonmat= yes doit # only the material plot, without the field: lena 1e-6 doit
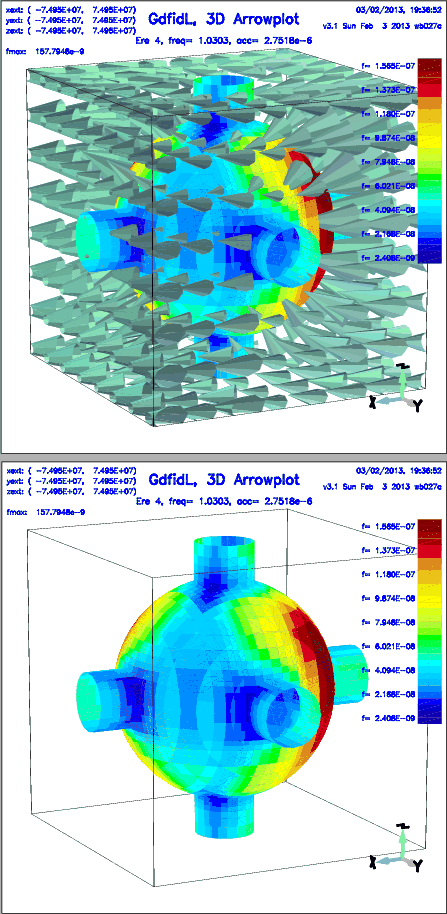 |
############################################################################## # Flags: nomenu, noprompt, nomessage, # ############################################################################## # Section: -lineplot # ############################################################################## # symbol = e_1 # # quantity = e # # solution = 1 # # component = z # # # # direction = z # # startpoint= ( 0.0, 0.0, -1.0e+30 ) # # used: ( undefined, undefined, undefined ) # ############################################################################## # plotopts = -geometry 690x560+10+10 # # showtext = yes -- (yes | no) # # onlyplotfiles= no -- (yes | no) # ############################################################################## # doit, ?, return, end, help, ls # ##############################################################################
symbol= QUAN_ISOL:
quantity= QUAN:
solution= ISOL:
component= [x|y|z|]:
direction= [x|y|z]:
startpoint= (X0, Y0, Z0):
plotopts= ANY STRING CONTAING OPTIONS FOR THE PLOT-PROGRAMS:
showtext= [yes|no]:
onlyplotfiles= [yes|no]:
doit:
-gen, i @last -linepl, sym e_3, comp x, dir z, sta (0,0,@zmin), doIn full glory, without abbreviations, this is:
-general
infile= @last
-lineplot
symbol= e_3
component= x
direction= z
startpoint= ( 0, 0, @zmin )
doit
############################################################################## # Flags: nomenu, noprompt, nomessage, # ############################################################################## # Section: -glineplot # ############################################################################## # symbol = e_1 # # quantity = e # # solution = 1 # # # # startpoint= ( 0.0, 0.0, -1.0e+30 ) # # direction = ( undefined, undefined, undefined ) # ############################################################################## # plotopts = -geometry 690x560+10+10 # # showtext = yes -- (yes | no) # # onlyplotfiles= no -- (yes | no) # ############################################################################## # @gvreal= undefined @gvimag= undefined @gvabs= undefined # ############################################################################## # doit, ?, return, end, help, ls # ##############################################################################
symbol= QUAN_ISOL:
quantity= QUAN:
solution= ISOL:
startpoint= (X0, Y0, Z0):
direction= ( Xn, Yn, Zn ):
plotopts= ANY STRING CONTAING OPTIONS FOR THE PLOT-PROGRAMS:
showtext= [yes|no]:
onlyplotfiles= [yes|no]:
doit:
direction will be plotted.
The component is plotted as a function of u.
The lineplot is performed a line
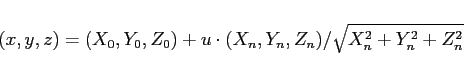
As a sideeffect, the complex voltage as seen by a particle is integrated.
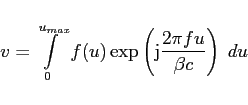
############################################################################## # Flags: nomenu, noprompt, nomessage, # ############################################################################## # Section: -2dplot # ############################################################################## # symbol = h_1 # # quantity = h # # solution = 1 # # # # component = z -- What Component to be plotted. # # normal = z -- The Plane Normal of the Cut-Plane. # # cutat = undefined -- The Coordinate of the Cut-Plane. # # ncontourlines= 30 -- The Number of Contourlines. # # fscale = auto # # # ############################################################################## # plotopts = -geometry 690x560+10+10 # # showtext = yes -- (yes | no) # # onlyplotfiles= no -- (yes | no) # ############################################################################## # doit, ?, return, end, help # ##############################################################################
symbol= QUAN_ISOL:
quantity= QUAN:
solution= ISOL:
component:
normal:
cutat:
ncontourlines:
fscale:
plotopts= ANY STRING CONTAING OPTIONS FOR THE PLOT-PROGRAMS:
showtext= [yes|no]:
onlyplotfiles= [yes|no]:
doit:
############################################################################## # Flags: nomenu, noprompt, nomessage, # ############################################################################## # Section: -energy # ############################################################################## # symbol = h_1 # # quantity = h # # solution = 1 # # bbxlow = -1.0000e+30, bbylow = -1.0000e+30, bbzlow = -1.0000e+30 # # bbxhigh= 1.0000e+30, bbyhigh= 1.0000e+30, bbzhigh= 1.0000e+30 # # # # # # @henergy : undefined (symbol: undefined, m: 1) # # @eenergy : undefined (symbol: undefined, m: 1) # ############################################################################## # doit, ?, return, end, help, ls # ##############################################################################
symbol= QUAN_ISOL:
quantity= QUAN:
solution= ISOL:
bbxlow=,bbxhigh=,bbylow=bbyhigh=,bbzlow=,bbzhigh=:
If the integrated field was an electric field, the result of the computation is stored in the symbolic variable @eenergy. If the integrated field was a magnetic field, the result of the compuation is stored in the symbolic variable @henergy.
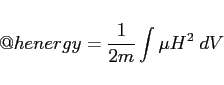 |
(2.1) |
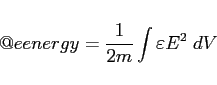 |
(2.2) |
m is 2 for resonant fields,
and 1 for nonresonant fields.
When you are computing resonant fields without periodic boundary conditions,
the fields that gd1.pp processes are the electric fields at a time t=0 and
the magnetic fields at a time t=T/4, where T=1/f. This means, you 'see' both,
the electric and magnetic fields at a time where they are at a maximum.
To get the total stored energy for resonant fields, one has to integrate
![]() over the volume at one instant
time. But E and H are not at the same time. They are offset by T/4.
The factor of 1/m, with m=2 for resonant fields, accounts for the effect of
integrating both the electric and magnetic fields at their (time) peak values.
over the volume at one instant
time. But E and H are not at the same time. They are offset by T/4.
The factor of 1/m, with m=2 for resonant fields, accounts for the effect of
integrating both the electric and magnetic fields at their (time) peak values.
When you are computing time dependent fields, the fields that gd1.pp
(normally) processes are electric and magnetic fields at (almost) the same
time (almost, because there is a time-offset of half of the used timestep, but
the used timestep is very small). For time dependent fields, the electric
and magnetic fields are not at their time-peak values, but more important,
the electric and magnetic fields are at the same time. The stored energy
can therefore be expressed directly by integrating
![]() over the volume.
No factor 'm' is needed.
over the volume.
No factor 'm' is needed.
You cannot and do not need to specify that 'm'. gd1.pp does this for you.
Example
To compute the stored energy in the first electric field found
in the database, we say:
-energy, symbol= e_1, doit
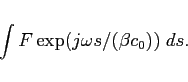
############################################################################## # Flags: nomenu, noprompt, nomessage, # ############################################################################## # Section: -lintegral # ############################################################################## # symbol = e_1 # # quantity = e # # solution = 1 # # # # direction = z # # component = z # # startpoint= ( 0.0, 0.0, -1.0e+30 ) # # (used) : ( @x0: undefined, @y0: undefined, @z0: undefined ) # # length = auto # # (@length) : undefined # # beta = 1.0 # # frequency = auto -- [auto | Real] # ############################################################################## # @vreal= undefined @vimag= undefined @vabs= undefined # ############################################################################## # doit, ?, return, end, help, ls # ##############################################################################
symbol= QUAN_ISOL:
quantity= QUAN:
solution= ISOL:
direction= [x|y|z]:
component= [x|y|z]:
startpoint= ( X0, Y0, Z0 ):
length:
auto, or a real number.
length= auto, the integral is performed from the startpoint
to the end of the computational domain (in direction-direction).
beta:
beta
as a very large number, say 1e+10.
frequency:
"auto", the frequency is taken from the dataset.
If frequency is specified as a number, that value is taken for
the integrand.
doit:
############################################################################## # Flags: nomenu, noprompt, nomessage, # ############################################################################## # Section: -wlosses # ############################################################################## # symbol = h_1 # # quantity = h # # solution = 1 # # frequency = auto -- [auto | Real] # # bbxlow = -1.0000e+30, bbylow = -1.0000e+30, bbzlow = -1.0000e+30 # # bbxhigh= 1.0000e+30, bbyhigh= 1.0000e+30, bbzhigh= 1.0000e+30 # # # # # # @metalpower : undefined [VA] (symbol: undefined) # ############################################################################## # doit, ?, return, end, help, ls # ##############################################################################The conductivities that are used in the perturbation formula may be entered in the section -material. The result of the computation is available as the symbolic variable @metalpower. The wall losses are computed as:
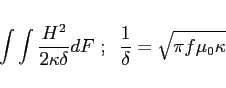
The integration is performed over all metallic surfaces that would appear in a plot as produced by the section -3darrowplot. This implies, that wall losses are NOT computed for electric symmetry planes, since the material on the symmetry planes are not shown in -3darrowplot.
symbol= QUAN_ISOL:
quantity= QUAN:
solution= ISOL:
frequency:
"auto", the frequency is taken from the dataset.
If frequency is specified as a number, that value is taken for
the integrand.
bbxlow=,bbxhigh=,bbylow=bbyhigh=,bbzlow=,bbzhigh=:
doit:
@metalpower for subsequent
calculations.
For electric fields:
![\begin{displaymath}
\frac{1}{m} \int \vec{E} \cdot \kappa_e \vec{E} \; dV = {\rm @epspower \; [VA]}
\end{displaymath}](img82.gif)
For magnetic fields:
![\begin{displaymath}
\frac{1}{m} \int \vec{H} \cdot \kappa_m \vec{H} \; dV = {\rm @muepower \; [VA]}
\end{displaymath}](img83.gif)
The integration is performed over the specified volume.
The time-averaging factor m is 2 for resonant fields,
and 1 for nonresonant fields.
############################################################################## # Flags: nomenu, noprompt, nomessage, # ############################################################################## # Section: -dlosses # ############################################################################## # symbol = h_1 # # quantity = h # # solution = 1 # # bbxlow = -1.0000e+30, bbylow = -1.0000e+30, bbzlow = -1.0000e+30 # # bbxhigh= 1.0000e+30, bbyhigh= 1.0000e+30, bbzhigh= 1.0000e+30 # # # # # # @muepower : undefined (symbol: undefined, m: 1) # # @epspower : undefined (symbol: undefined, m: 1) # ############################################################################## # doit, ?, return, end, help, ls # ##############################################################################
symbol= QUAN_ISOL:
quantity= QUAN:
solution= ISOL:
frequency:
"auto", the frequency is taken from the dataset.
If frequency is specified as a number, that value is taken for
the integrand.
bbxlow=,bbxhigh=,bbylow=bbyhigh=,bbzlow=,bbzhigh=:
doit:
@epspower or @muepower
for subsequent calculations.
############################################################################## # Flags: nomenu, noprompt, nomessage, # ############################################################################## # Section: -flux # ############################################################################## # symbol = h_1 # # quantity= h # # solution= 1 # # # # normal = z -- [x|y|z] # # cutat = undefined -- Coordinate of the integration Plane # # xlow= undefined , xhigh= undefined # # ylow= undefined , yhigh= undefined # # zlow= undefined , zhigh= undefined # # ( used: @cutat : undefined ) # # ( used: @xlow : undefined , @xhigh : undefined ) # # ( used: @ylow : undefined , @yhigh : undefined ) # # ( used: @zlow : undefined , @zhigh : undefined ) # # @flux : undefined [undefined] # ############################################################################## # doit, ?, return, end, help, ls # ##############################################################################
symbol= QUAN_ISOL:
quantity= QUAN:
solution= ISOL:
normal= [x|y|z]:
cutat= :
"normal" coordinate of the area through which the integration
shall be performed.
xlow= ??, xhigh= ??, ylow= ??, yhigh= ??, zlow= ??, zhigh= ??:
normal= x, the values xlow= ??, xhigh= ?? are ignored.
normal= y, the values ylow= ??, yhigh= ?? are ignored.
normal= z, the values zlow= ??, zhigh= ?? are ignored.
doit, the actually used values will be accessible as the symbolic
variables @cutat, @xlow, @xhigh, @ylow, @yhigh, @zlow, @zhigh.
doit:
@flux for subsequent
calculations.
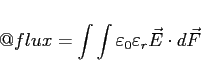
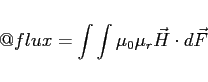
############################################################################## # Flags: nomenu, noprompt, nomessage, # ############################################################################## # Section: -clouds # ############################################################################## # symbol = h_1 # # quantity= h # # solution= 1 # # # # position = no -- 3D Plot of Particle Positions # # showbox= yes -- Show computational Box with 3D Plot # # gamma = yes -- 1D Plot of Gamma # # hgamma = yes -- 1D Plot of Gamma-density # # ihgamma= 30 -- Number of hgamma Bins # # normal = z -- Direction of 1D Plots (gamma) # # phase = yes -- Plots of velocities vs pos # # export = no -- Export raw Cloud Data # # outfile= ./gdfidl-exported-clouds # ############################################################################## # plotopts = -geometry 690x560+10+10 # # showtext = yes -- (yes | no) # # onlyplotfiles= no -- (yes | no) # ############################################################################## # doit, ?, return, end, help, ls # ##############################################################################
symbol= QUAN_ISOL:
quantity= QUAN:
solution= ISOL:
position= [yes|no]:
showbox= [yes|no]:
gamma= [yes|no]
hgamma= [yes|no]
ihgamma= NUMBER
hgamma= yes, specifies how many bins of average gamma shall be
used for the hgamma-plot. The total normal-extension of the
computational volume will be divided in ihgamma=NUMBER equal
sections.
The average of the gamma of all clouds within the bins will be plotted.
normal= [x|y|z]
hgamma plot will be produced.
phase= [yes|no]
export= [yes|no]
outfile=WHATEVER.
plotopts= ANY STRING CONTAING OPTIONS FOR THE PLOT-PROGRAMS:
showtext= [yes|no]:
onlyplotfiles= [yes|no]:
doit
############################################################################## # Flags: nomenu, noprompt, nomessage, # ############################################################################## # Section -sparameter # ############################################################################## # ports = all # # -- (all | LIST ) # # modes = ( 1 ) -- (all | LIST ) # # timedata = no -- ( yes | no ) # # tsumpower= no -- ( yes | no ) # # tintpower= no -- ( yes | no ) # # usample = 5 -- Undersample for t-Plots # # freqdata = yes -- ( yes | no ) # # wantdf = auto -- ( auto | REAL ) # # windowed = yes -- ( yes | no ) # # edgeofwindow= 0.70 -- At what Frac. start to apply Window # # fsumpower= yes -- ( yes | no ) # # magnitude= yes -- ( yes | no ) # # slog = no -- ( yes | no ) # # fintpower= no -- ( yes | no ) # # phase = no -- ( yes | no ) # # smithplot= no -- ( yes | no ) # # markerat= undefined -- Want a Marker at .. in Smith-Chart # # groupvelocity= no -- ( yes | no ) # # fri = no -- ( yes | no ) # # excdata = yes -- Show the Data of the Excitation # # upto = auto -- [s] ( auto | REAL ) # # tfirst = 0.0 -- [s] # # flow = auto -- [1/s] ( auto | REAL ) # # fhigh = auto -- [1/s] ( auto | REAL ) # # ignoreexc = no -- ( yes | no ) # # details = no -- ( yes | no ) # ############################################################################## # plotopts= -geometry 690x560+10+10 # # showtext = yes -- (yes | no) # # onlyplotfiles= no -- (yes | no) # ############################################################################## # return, help, end, ls, clearmarkers, doit # ##############################################################################
ports:
all, or a list of names of ports.
ports= all, the time dependent data of all ports found in the
data-base are processed.
ports is a list of names, only the time dependent data of the ports
whose names are found in the list are processed.
modes:
all, or a list of mode-numbers.
modes= all, the time dependent data of all modes of the selected
ports found in the data-base are processed.
modes is a list of mode-numbers, only the time dependent data of
the modes with numbers in the list are processed.
timedata:
timedata= yes, the time dependent amplitudes of the selected
modes are plotted.
tsumpower:
tintpower:
usample= NN:
freqdata:
freqdata= no, no scattering parameters are plotted.
wantdf= DF:
wantdf=auto, the computed scattering parameters have a
frequency resolution of just df = 1 / simulated time.
wantdf=DF , the time data are assumed to be zero outside
of the FDTD-simulated time window. These zero-padded time signals within
a time window of DT = 1 / DF are FFTed, giving an apparent frequency
resolution
of DF.
windowed:
edgeofwindow= FRAC:
![\begin{displaymath}\frac{1}{2} \left[1+\cos(\pi\frac{t-FRAC*T}{(1-FRAC)*T})\right] \end{displaymath}](img86.gif)
fsumpower:
magnitude:
magnitude= yes, the absolute value of the scattering parameters
are plotted.
slog:
slog= yes, the magnitude of the scattering parameters are
initially plotted in a logarithmic plot.
fintpower:
phase:
phase= yes, the phase of the scattering parameters are plotted.
smithplot:
smithplot= yes, the scattering parameters are plotted in a
smith-plot.
markerat= Fi:
groupvelocity:
groupvelocity= yes, the groupvelocity is computed from the
phase. The result only makes sense for transmissions.
fri:
fri= yes, the scattering parameters are written in fri-format
to files.
excdata:
upto:
auto or a time-value.
upto= auto, the time data up to the last found simulated time
are considered for the fourier-transforms.
upto= TMAX, only the time-data upto the time-value TMAX
are considered for the fourier-transforms. This is useful,
for the case that a late time instability of the time
domain computation has occured, and you want to know the scattering
parameters of a shorter simulation.
THIS SHOULD NOT HAPPEN.
IF YOU ENCOUNTER A LATE TIME INSTABILITY, PLEASE SEND THE INPUTFILE
WHERE THIS INSTABILITY HAS OCCURED TO "bruns@gdfidl.de".
wantdf= DF.
tfirst= TFIRST:
TFIRST are discarded and replaced by Zero.
flow:
flow= auto, the lower boundary of the frequency range of the
plots is derived from the centerfrequency and the bandwidth of the
excitation as they were specified in the input for
gd1.
If flow= FMIN, the lower boundary of the frequency range is
FMIN.
fhigh:
fhigh= auto, the upper boundary of the frequency range of the
plots is derived from the centerfrequency and the bandwidth of the
excitation as they were specified in the input for
gd1.
If fhigh= FMAX, the lower boundary of the frequency range is
FMAX.
ignoreexc:
ignoreexc= yes, the spectrum of the excited mode is ignored.
Instead, a flat spectrum is assumed.
This is useful e.g. to compute the coupling from an relativistic charge
to the port-modes.
details:
yes, a lot of intermediate results are plotted.
plotopts= ANY STRING CONTAING OPTIONS FOR THE PLOT-PROGRAMS:
showtext= [yes|no]:
onlyplotfiles= [yes|no]:
doit:
clearmarkers:
clearmarkers, the list of markers which were requested via
markerat is cleared.
Input, Output, we say:
-sparameter
modes= ( 1, 2 )
ports= ( Input, Output )
doit
gd1 < /usr/local/gd1/examples-from-the-manual/arndt.MTT90.p1854.fig5.gdf
# /usr/local/gd1/examples-from-the-manual/arndt.MTT90.p1854.fig5.gdf
define(INF,10000.0)
define(MAG, 2) define(PEL, 1) define(CU, 3) define(PER,0)
define(FREQ,15e9)
define(WGH, 15.8e-3)
define(WGW0, 7.9e-3)
define(T0, 6.6e-3)
define(CSW1, 4.2e-3)
define(T1, 6.2e-3)
define(STPSZE, 0.5e-3 )
define(XHIGH, 30e-3) define(XLOW, -XHIGH )
define(ZHIGH, 12e-3) define(ZLOW, -ZHIGH )
########
########
########
-general
outfile= /tmp/UserName/arndt
scratch= /tmp/UserName/arndt-scratch-
-mesh
spacing= STPSZE
# graded= on, qfgraded= 1.2, dmaxgraded= 3e8/FREQ/20
pxlow= XLOW, pxhigh= XHIGH
pylow= -WGH/2-STPSZE, pyhigh= 0 # Weg damit
pylow= 0, pyhigh= WGH/2+STPSZE
pzlow= ZLOW, pzhigh= ZHIGH
cxlow= el, cxhigh= el
cylow= el, cyhigh= mag # Weg damit
cylow= mag, cyhigh= ele
czlow= el, czhigh= mag
-material
material= PEL, type= electric
material= CU, type= electric
#
# Fill everything with material CU
#
-brick
material= CU,
volume= ( -INF, INF, \
-INF, INF, \
-INF, INF)
doit
#
# Upper and lower waveguide
#
material= 0,
volume= ( -INF,INF, -WGH/2,WGH/2, T0/2,T0/2+WGW0 ), doit
vol (-INF,INF, -WGH/2,WGH/2, -(T0/2+WGW0),-T0/2 ), doit
#
# coupling slits
#
vol ( -CSW1/2,CSW1/2, -WGH/2,WGH/2, -(T0/2+WGW0),T0/2+WGW0 ), doit
vol ( -(4.2e-3/2+3.0e-3),\
(4.2e-3/2+3.0e-3),\
-WGH/2,WGH/2,\
T0/2,T0/2+WGW0 ),
doit
vol ( -(4.2e-3/2+3.0e-3),4.2e-3/2+3.0e-3, -WGH/2,WGH/2, -(T0/2+WGW0),-T0/2 ), do
define(XDUM, 4.2e-3/2+3.0e-3 )
vol ( -(XDUM+3.2e-3),-XDUM, -WGH/2,WGH/2, -(T0/2+WGW0),T0/2+WGW0 ), do
vol ( XDUM,XDUM+3.2e-3, -WGH/2,WGH/2, -(T0/2+WGW0),T0/2+WGW0 ), do
define(XDUM, 4.2e-3/2+3.0e-3+3.2e-3+4.2e-3 )
vol ( -(XDUM+1.6e-3),-XDUM, -WGH/2,WGH/2, -(T0/2+WGW0),T0/2+WGW0 ), do
vol ( XDUM,XDUM+1.6e-3, -WGH/2,WGH/2, -(T0/2+WGW0),T0/2+WGW0 ), do
-volumeplot,
# eyeposition= ( 1.0, 2.0, 0.5)
scale= 4.5
doit
-fdtd
-ports
name= xlow1, plane= xlow, pzlow = 0, modes= 1, doit
name= xlow2, plane= xlow, pzhigh= 0, modes= 1, doit
name= xhigh1, plane= xhigh, pzlow = 0, modes= 1, doit
name= xhigh2, plane= xhigh, pzhigh= 0, modes= 1, doit
-pexcitation
port= xlow1,
mode= 1, amplitude= 1, frequency= 15e9, bandwidth= 10e9,
-time
tmin= 100/FREQ
tmax= 10000/FREQ
firsts= 10/FREQ
lasts = 11/FREQ
distancesave= 0.10/FREQ
amptresh= 1e-3
-fdtd
doit
In the next step, we start gd1.pp to compute the scattering parameters
from the time dependent amplitudes, that were computed by gd1.
The input we give to gd1.pp is:
-gen, inf @last
-3da, sy e_1, scal 4.5, arr 5000, doit
-spa, window yes, doit
window no, doit
The resulting plots are shown in the figures
2.2 to 2.12.
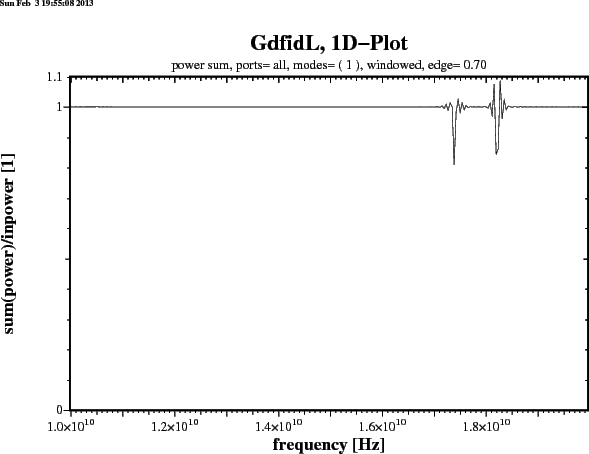 |
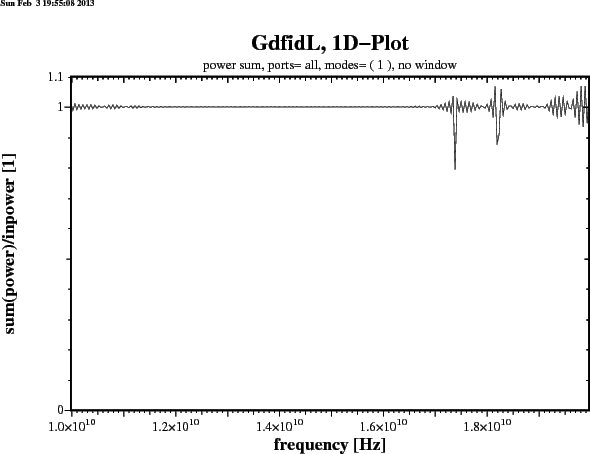 |
tfirst.
A Computation with a relativistic Line Charge is performed.
The Device is a Button-Beam-Position-Monitor. Only a Quarter of the Device is modeled.
There are three Ports where Energy can flow away from the Button.
The TEM-Line connected to the Button, and the lower and upper Beam-Pipe.
The Amplitudes at these Ports is recorded when modes at these Ports is specified
larger than Zero.
gd1 < /usr/local/gd1/examples-from-the-manual/Button-LineCharge.gdf
# /usr/local/gd1/examples-from-the-manual/Button-LineCharge.gdf
define(INF, 10000)
define(SIGMA, 4.6e-3)
define(STPSZE, 0.15e-3 )
define(A, 40e-3)
define(B, 37e-3)
define(ZYL1, 100e-3)
define(RADO, 3.8e-3)
define(RADI, 3.5e-3)
define(FEED, 0.33e-3)
define(BUTTON,4.0e-3)
define(CERMA, 4.5e-3)
define(CERME, 6.5e-3)
define(BLOCK, 7.5e-3)
define(EXT, 85e-3)
###############################################################
-general
outfile= /tmp/UserName/Resultfiles
scratch= /tmp/UserName/scratch-
-mesh
spacing= STPSZE
graded= yes, dmaxgraded= 4*STPSZE
define(BUTTONX0, 9.065e-3)
define(X1, BUTTONX0-RADO)
define(X2, BUTTONX0+RADO)
define(NX, 1+(X2-X1)/STPSZE)
xfixed( NX, X1, X2 )
zfixed( NX, -RADO, +RADO )
pxlow= 0, pxhigh= EXT+5e-3
pylow= 0, pyhigh= 25e-3
pzlow = -0.01
pzhigh= +0.01
cxlow= mag, cxhigh= ele
cylow= mag, cyhigh= ele
czlow= el, czhigh= ele
#########################################################################
-material
material= 3, type= electric
material= 4, type= electric
material= 5, type= normal, epsr= 9, muer= 1
-brick
# Fill the universe with metal..
material=4
volume= (-INF,INF, -INF,INF, -INF,INF)
doit
################################################################
#vacuum chamber
-ggcylinder
material= 0
originprime= ( 0, 0, 0 )
xprimedirection= ( 1, 0, 0 )
yprimedirection= ( 0, 1, 0 )
range= (-ZYL1/2, ZYL1/2)
show= later
clear
point= ( -36e-3, -5e-3)
point= ( -36e-3, 5e-3)
point= (-20.415e-3, 14e-3)
point= ( 20.415e-3, 14e-3)
point= ( 36e-3, 5e-3)
point= ( 56e-3, 5e-3)
point= ( 56e-3, 14e-3)
point= ( EXT, 14e-3)
point= ( EXT, -14e-3)
point= ( 56e-3, -14e-3)
point= ( 56e-3, -5e-3)
point= ( 36e-3, -5e-3)
point= ( 20.415e-3, -14e-3)
point= (-20.415e-3, -14e-3)
point= ( -36e-3, -5e-3)
doit
#
# All four Buttons are modelled, although
# only one of them is inside the selected Quarter.
#
do i= -1, 1, 2
do j= -1, 1, 2
#The vacuum part containing the bpm's
-gbor
material= 0
originprime= ( j * BUTTONX0, 14e-3*i, 0 )
zprimedirection= ( 0, i, 0 )
rprimedirection= ( 1, 0, 0 )
range= (0, 360)
show= later
clear
point= (0, 0.)
point= (0, 4.25e-3)
point= (27e-3, 4.25e-3)
point= (27e-3, 0.)
doit
# the bpm's
#the outside shell _1
material= 3
clear
point= (0, RADO)
point= (0, 4.25e-3)
point= (17.5e-3, 4.25e-3)
point= (17.5e-3, 4.25e-3)
point= (27e-3, 4.25e-3)
point= (27e-3, 2.05e-3)
point= (BLOCK, 2.05e-3)
point= (BLOCK, RADO)
point= (0, RADO)
doit
#inner part of the bpm's
clear
point= (0, 0.0)
point= (0, RADI)
point= (BUTTON, RADI)
point= (BUTTON, 1.65e-3)
point= (CERMA, 1.65e-3)
point= (CERMA, FEED)
point= (CERME, FEED)
point= (CERME, 0.89e-3)
point= (27e-3, 0.89e-3)
point= (27e-3, 0.0e-3)
doit
#Al203 part_1
material= 5
clear
point= (CERMA, FEED)
point= (CERMA, RADO)
point= (CERME, RADO)
point= (CERME, FEED)
doit
end do
end do
###############################################
-volumeplot
scale=3
doit
##########################################################################
#
# Wake-Parameters..
#
# For Ports where the Beam passes through, npml=40 or larger is recommended.
#
-fdtd
-ports
name= lower_end, plane= zlow, modes= 10, npml= 40, doit
name= upper_end, plane= zhigh, modes= 10, npml= 40, doit
name= bpmho, plane= yhigh, modes= 1, doit
-lcharge
xpos= 0, ypos= 0,
charge= 1e-12, sigma= SIGMA
shigh= 2
#
# The Storing of the Fields is just for the nice plot for the Manual.
#
-time
firstsaved= SIGMA / @clight
lastsaved= (ZYL1 + 20 * SIGMA) / @clight
distance= SIGMA / @clight
-fdtd
doit
The time Data of the Modes at the lower and upper Beam-Pipes are contaminated by the Passing of
the Beam through these Ports at small time Values.
The Beam has significant Charge ie more than 1e-7 than the Maximum, for times less
than
-general,
infile= @last
scratch= ./Resultfile-
-3darrow, sy e_11, arr 1e4, lena 100, fonmat yes, scale 4, doit
-spa,
freqdata= no, timedata= yes, tintpower= yes, modes= all
ports= ( lower_end )
tfirst= 0, doit
tfirst= 8 * SIGMA / 3e8, doit
ports= ( upper_end )
tfirst= 0, doit
tfirst= ( @zmax - @zmin + 8 * SIGMA ) / 3e8, doit
ports= ( bpmho )
tfirst= 0, doit
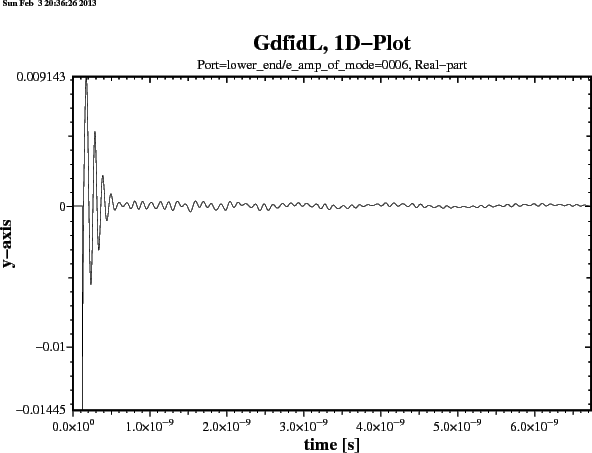 |
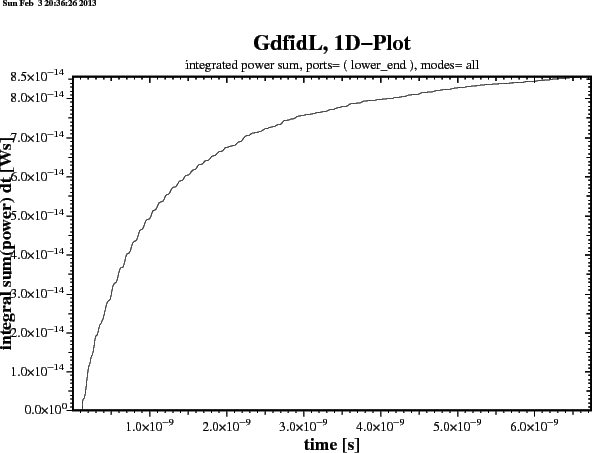 |
############################################################################## # Flags: nomenu, noprompt, nomessage, # ############################################################################## # section -material # ############################################################################## # material= 1 epsr....: infinity kappa = 58.0e+6 # # type: electric xepsr: infinity xkappa = 58.0e+6 # # yepsr: infinity ykappa = 58.0e+6 # # zepsr: infinity zkappa = 58.0e+6 # # muer....: 0.0 mkappa : 0.0 # # xmuer: 0.0 xmkappa: 0.0 # # ymuer: 0.0 ymkappa: 0.0 # # zmuer: 0.0 zmkappa: 0.0 # ############################################################################## # return, help, ls # ##############################################################################
type, epsr, muer, mkappa cannot be changed.
-wlosses
shall use a conductivity of
-material
material= 10
kappa= 30e6
gd1 computes the integral of the ![]() component along the outermost
paths where a beam can travel and stores the result in the database.
Since from these data the longitudinal and transverse wakepotentials everywhere
in the beam pipe can be computed, you can specify an unlimited number of
positions (x,y) where you are interested in the wakepotentials.
The (x,y) position of the exciting charge cannot be changed afterwards,
though.
component along the outermost
paths where a beam can travel and stores the result in the database.
Since from these data the longitudinal and transverse wakepotentials everywhere
in the beam pipe can be computed, you can specify an unlimited number of
positions (x,y) where you are interested in the wakepotentials.
The (x,y) position of the exciting charge cannot be changed afterwards,
though.
############################################################################## # Flags: nomenu, noprompt, nomessage, # ############################################################################## # section -wakes # ############################################################################## # set = 0 -- What Dataset (windowwake only) # # watq = yes -- Process all Wakes at Positions of Line-Charges # # awtatq = yes -- Use the Average of the two nearest transverse Wakes # # -- as the transverse Wakes at the Positions of Charges # # impedances= no -- Compute Impedances. # # wantdf = auto -- Wanted freq Resolution => SIN(f)/f Interpol. # # window = yes -- Apply Hann-Window when computing Impedances. # # fhigh = undefined -- fhigh of Impedances. # # uselu = yes -- Use Sparse LU-Factorisation (not MG). # # niter = 2 -- nIter for MG # # omega = 1.10 -- Omega for MG # # Omega2= 1.10 -- Omega2 for MG # # mgdetails= no -- Show Number of its etc # # uselowpass= no -- Use lowpass Filtering. # # usehighpass= no -- Use highpass Filtering. # # showchargemax= no -- Show Value of Charge Maximum. # # centerbunch= yes -- Shift Data. Bunch Center will be at s=0. # # xyref = ( 0.0, 0.0) -- refpoint for ??atxy-Data # # usexyref = no -- Use refpoint? # # clear -- Clears all the "w*at*" Values. # # watxy = ( undefined, undefined) -- want wz(xi,yi,s), i= 1 # # wxatxy= ( undefined, undefined) -- want wx(xi,yi,s), i= 1 # # wyatxy= ( undefined, undefined) -- want wy(xi,yi,s), i= 1 # # watsi = undefined -- want w(x,y,si), i= 1 # # watxi = undefined -- want w(xi,y,s), i= 1 # # liny= 20 -- Number of Lines in y-Direction. # # watyi = undefined -- want w(x,yi,s), i= 1 # # linx= 20 -- Number of Lines in x-Direction. # # wxatxi= undefined -- want wx(xi,y,s), i= 1 # # wxatyi= undefined -- want wx(x,yi,s), i= 1 # # wyatxi= undefined -- want wy(xi,y,s), i= 1 # # wyatyi= undefined -- want wy(x,yi,s), i= 1 # # istrides= 3 -- Distance of s-Points of the Plots # # -- in Units of "ds". # # slow = 0.0 -- Lowest s-Value to consider. # # shigh= undefined -- Highest s-Value to consider. # # watsfiles = -none- # xlowwats = -1.0e+30 # # xhighwats= 1.0e+30 # # ylowwats = -1.0e+30 # # yhighwats= 1.0e+30 # # frequency= undefined ( @ufrequency : undefined ) # # ( @zxesrf: undefined ) # # ( @zxesrf: undefined ) # # ( @zxesrf: undefined ) # # ( @xloss : undefined ) [VAs] # # ( @yloss : undefined ) [VAs] # # ( @zloss : undefined ) [VAs] # # ( @charge: undefined ) [As] # ############################################################################## # plotopts= -geometry 690x560+10+10 # # showtext = yes -- (yes | no) # # onlyplotfiles= no -- (yes | no) # ############################################################################## # return, help, end, clear, doit # ##############################################################################
-windowwake, one can compute
in a single run the wakepotentials of different numbers of periodic sections
of a periodic geometry. The different results are call sets. The number of
the set to analyse is specified.
If awtatq= yes, gd1.pp computes the transverse wakepotentials at the two positions between the grid planes that are nearest to the position of the exciting charge. The average of the two potentials are then assumed to be the transverse wakepotential at the position of the charge.
uselu=no, the linear equations are solved with
an iterative scheme, which requires less memory, but much more time.
usexyref=yes, the transverse wakepotentials at (XREF,YREF)
are computed, and the other transverse wakepotentials are
plotted as eg.
You can specify an unlimited number of (x,y)-positions where you want to know the longitudinal or transverse wakepotentials.
You can specify an unlimited number of s-positions where you want to know the longitudinal or transverse wakepotentials.
You can specify an unlimited number of y- or y-positions where you want to know the longitudinal wakepotentials.
If you specify wxatxi= Xi, the transverse wakepotential in x-direction will be plotted in the y-s plane at the x-coordinate between meshplanes nearest to Xi.
If you specify wxatyi= Yi, the transverse wakepotential in x-direction will be plotted in the x-s plane at the y-coordinate between meshplanes nearest to Yi.
If you specify wyatxi= Xi, the transverse wakepotential in y-direction will be plotted in the y-s plane at the x-coordinate between meshplanes nearest to Xi.
If you specify wyatyi= Yi, the transverse wakepotential in y-direction will be plotted in the x-s plane at the y-coordinate between meshplanes nearest to Yi.
You can specify an unlimited number of y- or y-positions where you want to know the transverse wakepotentials.
xlowwats etc the border planes of the region where w(x,y,s) shall
be written to wats-files.
plotopts= ANY STRING CONTAING OPTIONS FOR THE PLOT-PROGRAMS:
showtext= [yes|no]:
onlyplotfiles= [yes|no]:
@zloss, @xloss, @yloss after
a wake-potential computation.
-wake
watxi= 1e-3
watxi= 2e-3
-sparameter may be used to generate the fri-files.
############################################################################## # Flags: nomenu, noprompt, nomessage, # ############################################################################## # section: -totouchstone # ############################################################################## # outfile= ./s-matrix.sXp # # ports= 2 # # normalise= no # # # # ij= (1,1) -- Index of S-Parameter. # # file= -none- # -- Filename of S-Parameter. # # factor= 1.0 -- Factor to apply. # # iport= 1, fcutoff= 0.0 # # ### # # Already specified Files: # # ij: (1,1), factor: 1.0, fcutoff: 0.0 # # file: -none- # ij: (1,2), factor: 1.0, fcutoff: 0.0 # # file: -none- # ij: (2,1), factor: 1.0, fcutoff: 0.0 # # file: -none- # ij: (2,2), factor: 1.0, fcutoff: 0.0 # # file: -none- ############################################################################## # clear, doit, ?, return, end, help, ls # ##############################################################################
outfile= VALID-FILENAME:
ports= NN:
normalise:
ij= (I, J):
file= NAME-OF-A-FRI-FILE:
factor= NUMBER:
wantdf= DF),
and must have the same frequency range.
#!/bin/sh
#
# First Step:
# Computation of the four rows of the scattering matrix.
# The results of each computation are stored in a different file.
#
# In the inputfile "arndt.00.x.gdf", the outfile is defined as
# outfile= /tmp/bruw1931/garbage/arndt-excitation-at-PEXC
#
#
# At each computation, a different port is excited.
#
# In the inputfile, the excitation is defined via:
# -pexcitation
# port= PEXC,
# mode= 1, amplitude= 1, frequency= 17e9, bandwidth= 20e9,
#
for PEXC in xlow1 xhigh1 xlow2 xhigh2
do
gd1 -DPEXC=$PEXC < arndt.00.x.gdf | tee out-excitation=$PEXC
done
#
# Fouriertransform, and generating fri-files.
# The names of the fri-files are defined via
#
# -general, scratch= /tmp/bruw1931/garbage/exc-at-$PEXC-
#
# Frequency resolution is 10 MHz (wantdf= 10e6)
#
for PEXC in xlow1 xhigh1 xlow2 xhigh2
do
gd1.pp << EOF
-general
infile= /tmp/bruw1931/garbage/arndt-excitation-at-$PEXC
scratch= /tmp/bruw1931/garbage/exc-at-$PEXC-
-sparameter
window= yes, edgeofwindow= 0.7
wantdf= 10e6
flow= 5e9, fhigh= 25e9
timedata= no, fsumpower= no, magnitude= no, phase= no, smithplot= no
fri= yes
onlyplotfiles= yes
doit
EOF
done
#
# Second step.
# Combining the rows of the scattering matrix to one TouchStone-file.
#
gd1.pp << EOF
-totouchstone
clear # file= - undefined -
ports= 4
sdefine(BASE, /tmp/bruw1931/garbage/exc-at)
sdefine(P1, xlow1) sdefine(P2, xhigh1)
sdefine(P3, xlow2) sdefine(P4, xhigh2)
ij= (1,1), file= [BASE]-[P1]-[P1]_out_1.fri
ij= (1,2), file= [BASE]-[P1]-[P2]_out_1.fri
ij= (1,3), file= [BASE]-[P1]-[P3]_out_1.fri
ij= (1,4), file= [BASE]-[P1]-[P4]_out_1.fri
ij= (2,1), file= [BASE]-[P2]-[P1]_out_1.fri
ij= (2,2), file= [BASE]-[P2]-[P2]_out_1.fri
ij= (2,3), file= [BASE]-[P2]-[P3]_out_1.fri
ij= (2,4), file= [BASE]-[P2]-[P4]_out_1.fri
ij= (3,1), file= [BASE]-[P3]-[P1]_out_1.fri
ij= (3,2), file= [BASE]-[P3]-[P2]_out_1.fri
ij= (3,3), file= [BASE]-[P3]-[P3]_out_1.fri
ij= (3,4), file= [BASE]-[P3]-[P4]_out_1.fri
ij= (4,1), file= [BASE]-[P4]-[P1]_out_1.fri
ij= (4,2), file= [BASE]-[P4]-[P2]_out_1.fri
ij= (4,3), file= [BASE]-[P4]-[P3]_out_1.fri
ij= (4,4), file= [BASE]-[P4]-[P4]_out_1.fri
outfile= /tmp/bruw1931/garbage/touchstone.s4p
doit
EOF
##############
############################################################################## # Flags: nomenu, noprompt, nomessage, # ############################################################################## # section: -combine # ############################################################################## # outfile= ./s-matrix.sXp # show= yes -- Give a Plot of the resulting # # -- Scattering Parameters. # # kdevice= 1, smatrix= -none- # kdevice= 2, smatrix= -none- # kdevice= 3, smatrix= -none- # p1ik= ( 1, 1 ), p2ik= ( 1, 2 ), factor= 1.0 # # fcutoff= 0.0, epsmue= 1.0, linelength= 0.0 # # rpi= 1, pik= ( 1, 1 ) # # #### # # Specified Port Connections: # # -- none so far -- # # #### # # Specified outer Ports: # # -- none so far -- # ############################################################################## # connect, assign, clear, doit, ?, return, end, help # ##############################################################################
outfile=
kdevice= NUMBER, smatrix= FILENAME
p1ik= ( I1, K1 ), p2ik= ( I2, K2 ), factor= FACTOR,
fcutoff= FC, epsmue= EPSMUE, linelength= LL, connect
The port I1 of device K1 is connected to the port I2 of device K2
via a line of length=LL.
The line has the parameters cutoff=FC, eps*mue=EPSMUE.
The damping of the line is FACTOR.
rpi= IRPI, pik= (I,K), assign
show= [yes|no]:
doit:
-combine
outfile= ./filter.s2p
kdevice= 1, smatrix= /tmp/UserName/ipart=1.s2p
kdevice= 2, smatrix= /tmp/UserName/ipart=2.s2p
kdevice= 3, smatrix= /tmp/UserName/ipart=1.s2p
# Connect the ports at the cutplanes.
# i: Port, k: Device
p1ik = (2,1), p2ik = (1,2), fcutoff= 3.2e9,
epsmue= 1, linelength= -5e-3, factor= 1, connect
p1ik = (2,2), p2ik = (2,3), fcutoff= 3.2e9,
epsmue= 1, linelength= -5e-3, factor= 1, connect
# rpi : Resulting Port I
# pik : Port I of device K
rpi= 1, pik= (1,1), assign
# The port '1' of the resulting device shall be the port '1' of device '1'.
rpi= 2, pik= (1,3), assign
# The port '2' of the resulting device shall be the port '1' of device '3'.
doit
-smonitor,
ie. scalar flux quantities.
############################################################################## # Flags: nomenu, noprompt, nomessage, # ############################################################################## # section: -smonitor # ############################################################################## # symbol = - none - # # timedata = yes -- ( yes | no ) # # upto = auto -- [s] ( auto | REAL ) # # freqdata = yes -- ( yes | no ) # # wantdf = auto -- ( auto | REAL ) # # windowed = yes -- ( yes | no ) # # edgeofwindow= 0.70 -- At what Frac. start to apply Window # # flow = auto -- [1/s] ( auto | REAL ) # # fhigh = auto -- [1/s] ( auto | REAL ) # ############################################################################## # plotopts= -geometry 690x560+10+10 # # showtext = yes -- (yes | no) # # onlyplotfiles= no -- (yes | no) # ############################################################################## # doit, ?, return, end, help, ls # ##############################################################################
symbol= BLA_BLA_BLUB:
timedata= [yes|no]:
upto= [auto|REAL]:
freqdata= [yes|no]:
wantdf= REAL:
windowed= [yes|no]:
edgeofwindow= REAL:
flow, fhigh : REAL:
plotopts= ANY STRING CONTAING OPTIONS FOR THE PLOT-PROGRAMS:
showtext= [yes|no]:
onlyplotfiles= [yes|no]:
doit:
############################################################################## # Flags: nomenu, noprompt, nomessage, # ############################################################################## # Section: -fexport # ############################################################################## # symbol = h_1 # # quantity= h # # solution= 1 # # phase = 45.0000 -- Only for Portmodes. # # bbxlow = -1.0000e+30, bbylow = -1.0000e+30, bbzlow = -1.0000e+30 # # bbxhigh = 1.0000e+30, bbyhigh= 1.0000e+30, bbzhigh= 1.0000e+30 # # outfile = ./gdfidl-exported-field # ############################################################################## # doit, ?, return, end, help, ls # ##############################################################################
symbol= QUAN_ISOL:
quantity= QUAN:
solution= ISOL:
bbxlow=,bbxhigh=,bbylow=bbyhigh=,bbzlow=,bbzhigh=:
outfile=:
doit:
-fexport or
gd1.pp's section -fexport can be used to create gif-files.
############################################################################## # Flags: nomenu, noprompt, nomessage, # ############################################################################## # Section: -2dmanygifs # ############################################################################## # 1stinfile = /tmp/UserName/fexported--000000000.gz # # outfiles = auto # # mpegfile = 2dmanygifs.mpeg # # uptonfiles= 1000000000 # # ixoffset = 0 # # iyoffset = 0 # # stride = 1 # # width = 1000 # # scale = 1.0 # # what = rHy -- rHx, rHy, Hx, Hy # # log = no -- plot log(1+|scale*f|) # # zerolines = yes -- Plot Lines at f=0 # # show = yes -- Show the mpeg. # ############################################################################## # doit, ?, return, end, help # ##############################################################################
1stinfile= FILENAME-NUMBER:
outfiles:
outfiles=auto,
the gif files will have the same name as the read fexported files, with
the letters .gif attached.
mpegfile:
uptonfiles=NFILES:
1stinfile=FILENAME,
and guess the next files from the name of the first file, ie. it will
count up the trailing number. It will read up to NFILES.
ixoffset=IX0, iyoffset= IY0, what= [rHx|rHy]:
what=rHx gives plots of the x-component of the field, multiplied by the
distance from the axis, what=rHy gives plots of the y-component of the
field, multplied by the distance from the axis.
doit:
-fexport, what=honmat can be used to create gif-files.
############################################################################## # Flags: nomenu, noprompt, nomessage, # ############################################################################## # Section: -3dmanygifs # ############################################################################## # 1stinfile = /tmp/UserName/H-onmat--000000001.gz # # outfiles = auto # # uptonfiles= 1000000000 # # what = abs -- abs, logabs, loglogabs # # mpegfile = ./3dmanygifs.mpeg # # xrot = -30.0 # # yrot = 40.0 # # zrot = 0.0 # # dxrot= 0.0 # # dyrot= 0.0 # # dzrot= 0.0 # # scale= 1.0 -- Plot scale*f. # # width= 1000 -- Width of the GIFs. # # boxed = yes -- Display Volume Box # ############################################################################## # doit, ?, return, end, help # ##############################################################################
1stinfile= FILENAME-NUMBER:
outfiles:
outfiles=auto,
the gif files will have the same name as the read fexported files, with
the letters .gif attached.
uptonfiles=NFILES:
1stinfile=FILENAME,
and guess the next files from the name of the first file, ie. it will
count up the trailing number. It will read up to NFILES.
what= [absh | logabs]:
mpegfile:
xrot= XROT, yrot= YROT, zrot= ZROT:
dxrot= DXROT, dyrot= DYROT, dzrot= DZROT:
boxed:
doit:
sdefine(name, value)
or a sequence
define(name, value).
sdefine(name, value),
the value of the variable may be up to 132 characters long.
It may contain any characters inside, except for '(\)'.
Leading and trailing blanks in the value are ignored.
define(name, expr),
the value of the variable is value of the arithmetic expression
expr.
gd1 -Dname=value.
This way, you can e.g. compute dispersion relations with simple shell scripts.
Variables defined in this way are not automatically evaluated.
#!/bin/sh
# Given the proper "inputfile.gdf", this shell-script computes the
# dispersion relation of some periodic structure.
for PHASE in 0 20 40 60 80 100 120 140 160 180
do
gd1 -DThisPhase=$PHASE < inputfile.gdf > out.Phase=$PHASE
done
eval(, the matching closing
brace is searched and the string inside the enclosing braces is interpreted
as an arithmetic expression. The value of the expression is transformed to
a string and substituted for eval(expression).
echo (2*3) # this outputs "(2*3)" echo eval(2*3) # this outputs "6"The arithmetic expression may contain the arithmetic operators
+,-,*,/,**,%.
In addition to that, the boolean operators ==,!=,<,>,<=,>= are handled.
The result of applying a boolean operator is an integer 0 or 1.
Zero stands for false, and 1 for true.
The functions
abs(x), min(x,y), max(x,y),
log(x), cos(x), sin(x), tan(x),
atan(x), atan2(x,y), mod(x,y), pow(x,y) are recognised and evaluated.
In every context, where a number is required as a parameter, an arithmetic
expression can be used. In these contexts, enclosing the expression in
eval() is not required.
do M1, M2, M3
# Loop-Body
enddo
The loop-body may itself contain do-loops, macro calls, whatever.
The iteration variable M1 is not restricted to integer values.
do i= 1, 100, 1 # count upwards
echo I is i
enddo
do i= 100, 1, -1 # count downwards
echo I is i
enddo
do i= 1, 2, 0.1 # non integer step
echo I is i
echo 2*I is eval(2*i)
enddo
if blocks.
if block is:
if (ARITHMETIC-EXPRESSION) then
#
# if-body
#
endif
If the ARITHMETIC-EXPRESSION evaluates to something else than '0'
then the body of the If-Block is executed.
A general if block is:
if (ARITHMETIC-EXPRESSION) then
#
# if-body
#
elseif (ARITHMETIC-EXPRESSION) then
#
# elseif-body
#
else
#
# else-body
#
endif
If-blocks may be nested.
macro followed by the name of the macro.
All lines until a line with only the keyword endmacro are considered
the body of the macro.
When gd1 or gd1.pp find such a macro, they read it and store the body of the
macro in an internal buffer.
#
# This defines a macro with name 'foo'
#
macro foo
echo I am foo, my first argument is @arg1
echo The total number of arguments supplied is @nargs
endmacro
When gd1 or gd1.pp find a call of the macro, the number of the supplied
arguments is assigned to the variable @nargs, and the variables
@arg1, @arg2, .. are assigned the values of the supplied parameters
of the call.
Similiar to the user definable variables (via sdefine),
the values of the arguments are strings. Of course, it is possible to have a
string, e.g. '1e-4', which happens
to be interpreted in the right context as a real number.
# # this calls 'foo' with the arguments 'hi', 'there' # call foo(hi, there)Macro calls may be nested. The body of a macro may call another macro.
@.
The exact name can be found in the description of the sections of gd1.pp.
There are some other variables as well that have not been described.
@pi, @clight:
These are the values of @nx, @ny, @nz: These contain the number of grid planes in the
three coordinate directions.
@x(i), @y(i), @z(i): These are the positions of the i.th gridplane.
@xmin, @xmax, @ymin, @ymax:
These are the extreme coordinates of the computational volume.
@path. Its value is a command string
that would enter the current section.
This section shows four macros that define a sequence of secondary
computations to be performed in the postprocessor gd1.pp.
All four macros are contained in the file
/usr/local/gd1/postprocessor-macros.
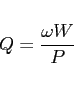 |
(4.1) |
-wlosses we compute the wall losses via the
perturbation formula. In the section -energy we compute the stored
energy in the h-field.
macro contains the commands to evaluate the above
formula for a given resonant field.
This macro is contained in the file /usr/local/gd1/postprocessor-macros.
macro QValue
pushflags, noprompt, nomenu, nomessage
define(QValue_PATH, @path) # remember current section
-base # goto the base of the branch-tree
-energy # compute stored energy
quantity= h # ... we dont need to compute the
solution= @arg1 # energy in the electric field
doit # -- it has to be the same
-wlosses # Wall-losses
doit
echo
echo *** h-Energy is @henergy
echo *** metalpower is @metalpower
return
define(QValue_value, eval(2*@pi*@frequency*2*@henergy/@metalpower))
echo *** mode number is @arg1
echo *** frequency is @frequency {Hz}
echo *** QValue is QValue_value {1}
# echo return path is : QValue_PATH
QValue_PATH # back to where we came from ...
undefine(QValue_PATH)
popflags
endmacro
With the definition of the macro available, we can compute the
Q-Value of the first resonant mode by saying:
call QValue(1)To compute the Q-values of the first five modes, we may say:
do i= 1, 5
call QValue(i)
enddo
macro perQValue
pushflags, noprompt, nomenu, nomessage
define(perQValue_PATH, @path) # remember current section
-base # goto the base of the branch-tree
-energy # compute stored energy
quantity= hre # ... we dont need to compute the
solution= @arg1 # energy in the electric field
doit # -- it has to be the same
# echo *** W_h of real part is @henergy
define(hre_energy, @henergy)
quantity= him
doit
define(him_energy, @henergy)
define(htot_energy, eval(hre_energy+him_energy) )
-wlosses # Wall-losses
quantity= hre, doit
define(hre_metalpower, @metalpower)
quantity= him, doit
define(him_metalpower, @metalpower)
define(htot_metalpower, eval(hre_metalpower+him_metalpower))
# echo *** total h-Energy is htot_energy
# echo *** total metalpower is htot_metalpower
define(perQValue_value, eval(2*@pi*@frequency*2*htot_energy/htot_metalpower))
echo
echo *** mode number is @arg1
echo *** frequency is @frequency {Hz}
echo *** QValue is perQValue_value {1}
# echo return path is : perQValue_PATH
perQValue_PATH # back to where we came from ...
undefine(perQValue_PATH)
popflags
endmacro
With the definition of the macro available, we can compute the
Q-Value of the first resonant mode by saying:
call perQValue(1)To compute the Q-values of the first five modes, we may say:
do i= 1, 5
call perQValue(i)
enddo
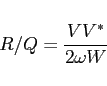 |
(4.2) |
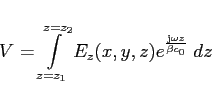 |
(4.3) |
macro contains the commands to evaluate the above
formula for a given resonant field.
This macro is contained in /usr/local/gd1/postprocessor-macros.
macro rshunt
pushflags, noprompt, nomenu, nomessage
define(rshunt_PATH, @path) # remember current section
-base # goto the base of the branch-tree
-energy # compute stored energy
quantity= e # ... we dont need to compute the
solution= @arg1 # energy in the magnetic field
doit # -- it has to be the same
# echo *** W_e is @eenergy
return
-lintegral # accelerating voltage
direction= z, component= z
startpoint= (0,0, @zmin)
length= auto
doit
# echo *** vabs is @vabs
return
define(rshunt_value_a, eval(@vabs **2/(2*@pi*@frequency*(2*2*@eenergy))))
define(rshunt_value_r, eval(@vreal**2/(2*@pi*@frequency*(2*2*@eenergy))))
define(rshunt_value_i, eval(@vimag**2/(2*@pi*@frequency*(2*2*@eenergy))))
echo
echo *** mode number is @arg1
echo *** frequency is @frequency {Hz}
echo ***
echo *** shunt impedances as computed from | U * conjg(U) | :
echo *** Shunt Impedance/Q is rshunt_value_a {Ohms}
echo *** Shunt Impedance/Q/m is eval(rshunt_value_a/@length) {Ohms/m}
echo ***
echo *** shunt impedances as computed from | Re(U) * Re(U) | :
echo *** Shunt Impedance/Q is rshunt_value_r {Ohms}
echo *** Shunt Impedance/Q/m is eval(rshunt_value_r/@length) {Ohms/m}
echo ***
echo *** shunt impedances as computed from | Im(U) * Im(U) | :
echo *** Shunt Impedance/Q is rshunt_value_i {Ohms}
echo *** Shunt Impedance/Q/m is eval(rshunt_value_i/@length) {Ohms/m}
## echo return path is : rshunt_PATH
rshunt_PATH # back to where we came from ...
undefine(rshunt_PATH)
## echo return path is : rshunt_PATH
popflags
endmacro
macro perrshunt
pushflags, noprompt, nomenu, nomessage
define(rshunt_PATH, @path) # remember current section
-base # goto the base of the branch-tree
-energy # compute stored energy
quantity= ere # ... we dont need to compute the
solution= @arg1 # energy in the magnetic field
doit # -- it has to be the same
# echo *** W_e of real part is @eenergy
define(ere_energy, @eenergy)
quantity= eim
doit
define(eim_energy, @eenergy)
define(etotenergy, eval(ere_energy+eim_energy) )
return
-lintegral # accelerating voltage
direction= z, component= z
startpoint= (0,0, @zmin)
length= auto
quantity= ere, doit
# echo *** vabs of real part is @vabs
define(V_ere_re, @vreal) define(V_ere_im, @vimag)
quantity= eim, doit
# echo *** vabs of imaginary part is @vabs
define(V_eim_re, @vreal) define(V_eim_im, @vimag)
return
define(vztotre, eval(V_ere_re-V_eim_im))
define(vztotim, eval(V_ere_im+V_eim_re))
define(vztotabs, eval((vztotre**2+vztotim**2)**0.5) )
define(rshunt_value, eval(vztotabs**2/(2*@pi*@frequency*(2*etotenergy))))
echo
echo *** mode number is @arg1
echo *** frequency is @frequency {Hz}
echo *** Shunt Impedance is rshunt_value {Ohms}
echo *** Shunt Impedance/m is eval(rshunt_value/@length) {Ohms/m}
# echo return path is : rshunt_PATH
rshunt_PATH # back to where we came from ...
undefine(rshunt_PATH)
popflags
endmacro
There is no example for computing eigenvalues, since this is mentioned in great detail in the tutorial.
The computation of wakepotentials occurs in two steps.
# /usr/local/gd1/examples-from-the-manual/wake-example-1.gdf
define(LargeNumber, 1000)
#
# The following picture shows a cut through the structure to be
# modelled and the variables associated to the lengths.
#
#
# a a a
# |<------------->|<-------------->|<----------->|
#
# - ------------------
# ^ | |
# b | | |
# | | |
# ----------------- --------------- -
# | beam | |
# |<-------------------------------------------->| | d
# | | |
# ------------------------------------------------ -
#
# x^
# |-> z
#
define( a, 1e-2 )
define( b, 5e-3 )
define( c, 5e-3 )
define( d, 1e-2 )
-general
outfile= /tmp/UserName/wake-example
scratch= /tmp/UserName/wake-example-scratch
text()= A strange geometry,
text()= it serves only as an example
text()= for computing wake-potentials.
-mesh
define(STPSZE, 3*a/60 )
spacing= STPSZE
perfectmesh= no
pxlow= 0, pxhigh= c+b
pylow= -STPSZE, pyhigh= d
pzlow= 0, pzhigh= 3*a
cxlow= ele, cxhigh= ele
cylow= ele, cyhigh= ele
czlow= ele, czhigh= ele
#
# We enforce a meshline at the position of the linecharge
# by enforcing two meshplanes
#
xfixed(1, c/2, 0)
yfixed(1, d/2, 0)
-brick
#
# fill the universe with metal
#
material= 1
volume= (-LargeNumber, LargeNumber,\
-LargeNumber, LargeNumber,\
-LargeNumber, LargeNumber)
doit
#
# carve out the waveguide
#
mat 0
xlow= 0, xhigh= c
ylow= 0, yhigh= LargeNumber
zlow= -LargeNumber, zhigh= LargeNumber
doit
#
# Carve out the resonator box
#
mat 0
xlow= 0, xhigh= c+b
ylow= 0, yhigh= LargeNumber
zlow= a, zhigh= 2*a
doit
-volumeplot
eyepos= ( 1.0, 2.30, 0.5 )
showlines= yes
scale= 2.5
doit
-fdtd
-ports
name= zlow, plane= zlow, modes= 0, doit
name= zhigh, plane= zhigh, modes= 0, doit
-lcharge
charge= 1 # 1 As
sigma= 5e-3
xposition= c/2
yposition= d/2
-fdtd
doit
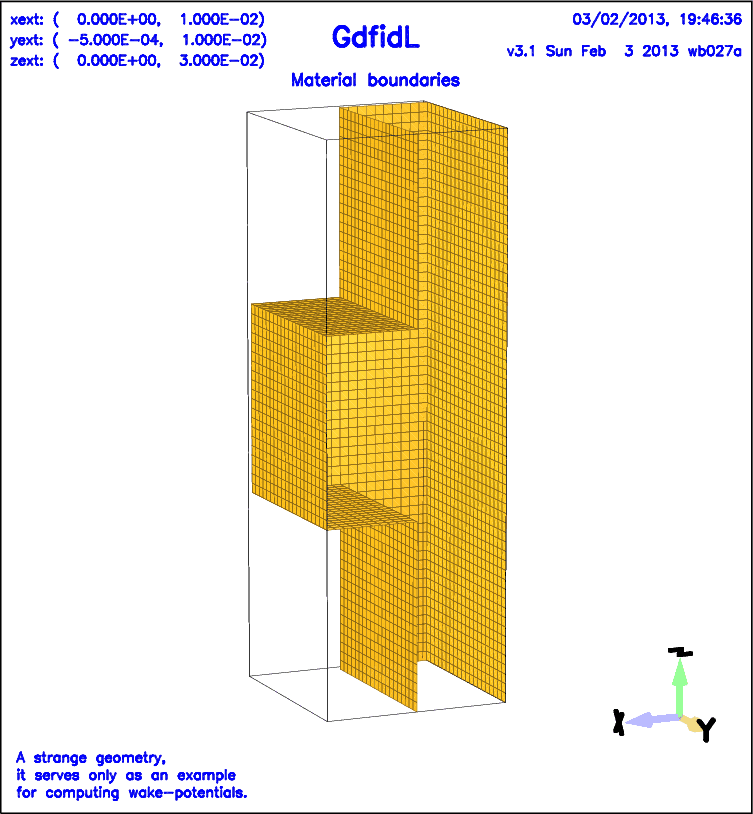 |
gd1 < wake-example-1.gdf | tee out
The next step is to tell the postprocessor that we wish to see the wakepotentials: The commands for the postprocessor gd1.pp are:
-general, infile= @last
-wakes
doit
We get three plots for the three components of the wakepotential at
the (x,y) coordinate where the line-charge was travelling.
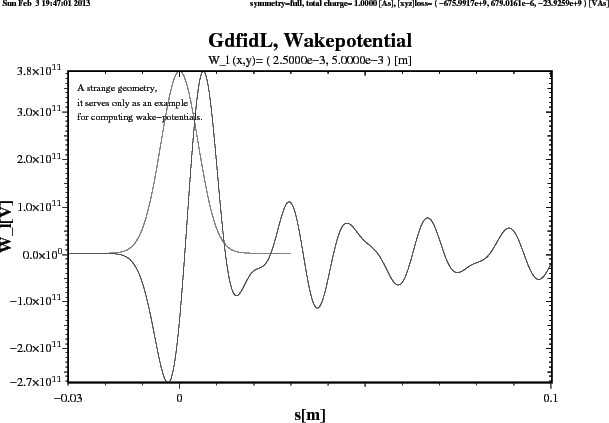 |
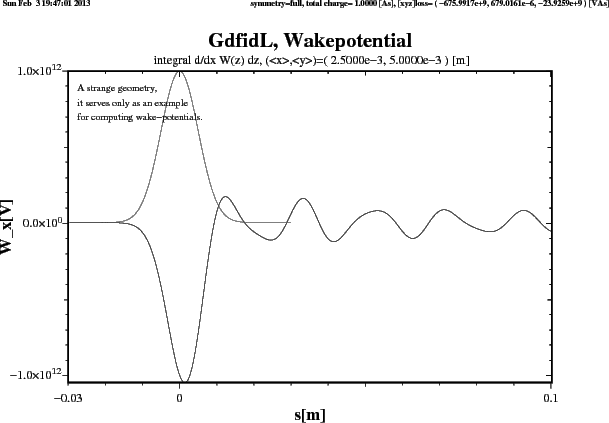
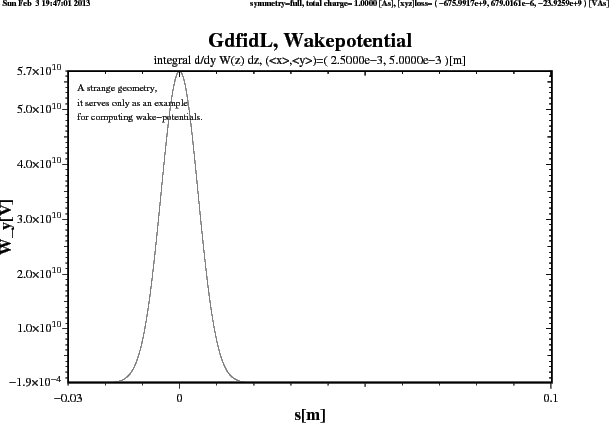 |
The computation of scattering Parameters is via in two Steps.
# /usr/local/gd1/examples-from-the-manual/spar-example-1.gdf
define(LargeNumber, 1000)
define( a, 1e-2 )
define( b, 5e-3 )
define( c, 5e-3 )
define( d, 1e-2 )
define(FREQ, 20e9)
-general
outfile= /tmp/UserName/spar-example
scratch= /tmp/UserName/spar-example-scratch
text()= A strange geometry, just an example
-mesh
define(STPSZE, 3*a/60 )
spacing= STPSZE
graded= yes, qfgraded= 1.2, dmaxgraded= @clight / FREQ / 20
perfectmesh= no
pxlow= 0, pxhigh= c+b
pylow= -STPSZE, pyhigh= d
pzlow= 0, pzhigh= 3*a
cxlow= ele, cxhigh= ele
cylow= ele, cyhigh= ele
czlow= ele, czhigh= ele
-brick
#
# fill the universe with metal
#
material= 1
volume= (-LargeNumber, LargeNumber,\
-LargeNumber, LargeNumber,\
-LargeNumber, LargeNumber)
doit
#
# carve out the waveguide
#
mat 0
xlow= 0, xhigh= c
ylow= 0, yhigh= LargeNumber
zlow= -LargeNumber, zhigh= LargeNumber
doit
#
# Carve out resonator box
#
mat 0
xlow= 0, xhigh= c+b
ylow= 0, yhigh= LargeNumber
zlow= a, zhigh= 2*a
doit
-volumeplot
eyepos= ( 1.0, 2.30, 0.5 )
showlines= yes
scale= 3
doit
-fdtd
-ports
name= Input, plane= zlow, modes= 1, doit
name= Output, plane= zhigh, modes= 1, doit
-pexcitation
port= Input
mode= 1
amplitude= 1
frequency= FREQ
bandwidth= 0.7*FREQ
-time
#
# tminimum: the minumum time to be simulated
# tmaximum: the maximum time to be simulated
# If the amplitudes have died down sufficiently
# at a time between tmin and tmax,
# the computation will stop.
#
tmin= 10/FREQ
tmax= 1000/FREQ
amptresh= 1e-3
-fdtd
doit
We start gd1 with the Command:
gd1 < spar-example-1.gdf | tee out
The next Step is to tell the Postprocessor that we wish the scattering Parameters to be computed and plotted. In addition to the default Values, we want to see (Parts of) the Timedata that were recorded during the Time-Domain Computation, and we want to see the scattering Parameters in a Smith-Chart. The Commands for the Postprocessor gd1.pp are:
-general, infile= @last
-sparameter
ports= all, modes= 1
timedata= yes
smithplot= yes, markerat 19e9, markerat 20e9, markerat 21e9
doit
We get in total 9 Plots.
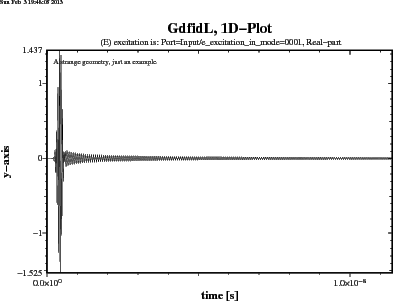
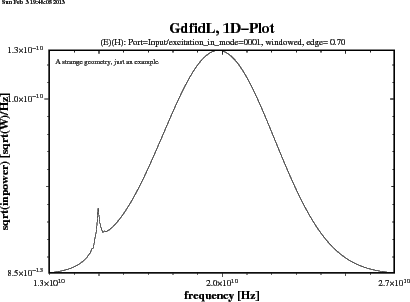 |
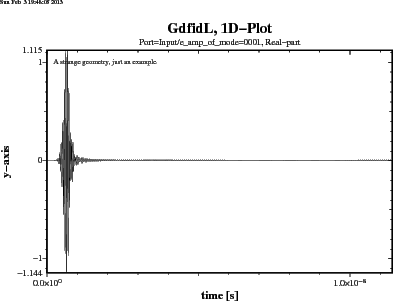
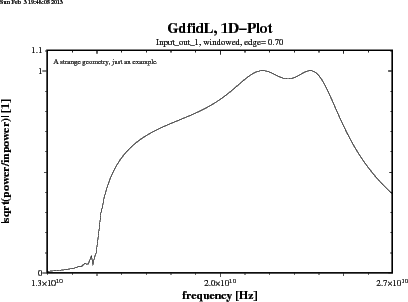
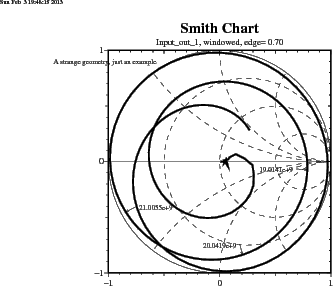 |
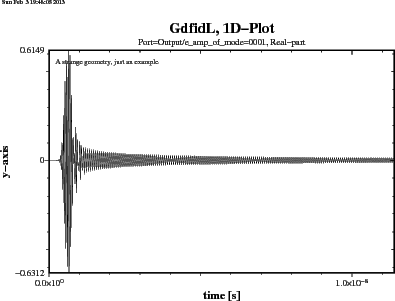
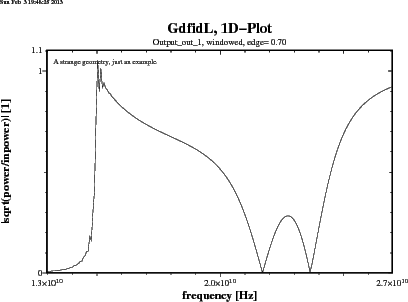
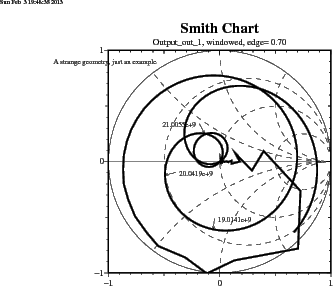 |
 |
Brillouin diagrams are plots of resonant frequencies as a function of the phase in periodic structures. GdfidL allows computation with specified phase shifts in x- y- and z-direction simultaneously. So we can compute Brillouin diagrams in 3D periodic structures, where the plane normals of the planes of periodicity are in x- y- and z-direction simultaneously.
The following inputfile defines an elemental cell of such a periodic structure.
# /usr/local/gd1/examples-from-the-manual/brillo.gdf
#
# Assign a value to "PHASE", if it is not yet defined
# via "gd1 -DPHASE=XX"
#
if (! defined(PHASE) ) then
define(PHASE, 45)
endif
#
# What part of the Brillouin-diagram do we want to compute?
#
#
# part==1 : from Gamma to H : 0<kx<pi/d, ky=0, kz=0
# part==2 : from H to N : kx=pi/d, 0<ky<pi/d, kz=0
# part==3 : from N to P : kx=pi/d, ky=pi/d, 0<kz<pi/d
# part==4 : from P to Gamma : 0 < (kx=ky=kz) < pi/d
#
#
# get the value of PART by inclusion of a file:
# the content of the file is simply
# "define(PART, 1)"
# or "define(PART, 2)"
# or "define(PART, 3)"
# or "define(PART, 4)"
include(this-part-of-brillo)
if ( PART == 1 ) then
define(XPHASE, PHASE)
define(YPHASE, 000)
define(ZPHASE, 000)
endif
if ( PART == 2 ) then
define(XPHASE, 180)
define(YPHASE, PHASE)
define(ZPHASE, 000)
endif
if ( PART == 3 ) then
define(XPHASE, 180)
define(YPHASE, 180)
define(ZPHASE, PHASE)
endif
if ( PART == 4 ) then
define(XPHASE, PHASE)
define(YPHASE, PHASE)
define(ZPHASE, PHASE)
endif
define(INF, eval(10000.0 *@clight))
define(MAG, 2) define(EL, 1)
##
## Geometry definitions
##
define(LATTICE_D, eval(@clight / 2) )
define(RADIUS, eval(LATTICE_D * 0.375) )
#
# default mesh spacing
#
define(STPSZE, eval(RADIUS/10))
##############
##############
##############
##############
##############
-general
outfile= /tmp/UserName/outfile
scratchbase= /tmp/UserName/delete-me-
text()= lattice constant d= LATTICE_D
text()= radius of the spheres= RADIUS
text()= r/d = eval(RADIUS/LATTICE_D)
text()= 2r/d = eval(2*RADIUS/LATTICE_D)
text()= stpsze= STPSZE
text()= xphase: XPHASE
text()= yphase: YPHASE
text()= zphase: ZPHASE
-mesh
spacing= STPSZE
graded= yes, dmaxgraded= 10*STPSZE
qfgraded= 1.3
perfectmesh= yes
perfectmesh= no
pxlow= -0.5*LATTICE_D, pxhigh= 0.5*LATTICE_D
pylow= -0.5*LATTICE_D, pyhigh= 0.5*LATTICE_D
pzlow= -0.5*LATTICE_D, pzhigh= 0.5*LATTICE_D
xperiodic= yes, xphase= XPHASE
yperiodic= yes, yphase= YPHASE
zperiodic= yes, zphase= ZPHASE
do ii= -1, 1, 1
xfixed( 2, ii*LATTICE_D-RADIUS, ii*LATTICE_D+RADIUS )
xfixed( 2, (ii-0.1)*LATTICE_D, (ii+0.1)*LATTICE_D )
yfixed( 2, ii*LATTICE_D-RADIUS, ii*LATTICE_D+RADIUS )
yfixed( 2, (ii-0.1)*LATTICE_D, (ii+0.1)*LATTICE_D )
zfixed( 2, ii*LATTICE_D-RADIUS, ii*LATTICE_D+RADIUS )
zfixed( 2, (ii-0.1)*LATTICE_D, (ii+0.1)*LATTICE_D )
enddo
##############
-brick
#
# Fill the universe with vacuum
#
material= 0
volume= (-INF,INF, -INF,INF, -INF,INF)
doit
define(M3, 1)
#
# define square lattice
# Only a part of these spheres end up being within
# computational volume
#
do iz= 0, 1, 1
do ix= 0, 1, 1
do iy= 0, 1, 1
#
# a sphere with center at
# ( ix*LATTICE_D, iy*LATTICE_D, iz*LATTICE_D )
#
-gbor
material= M3
origin= ( ix*LATTICE_D, \
iy*LATTICE_D, \
iz*LATTICE_D )
rprimedirection= ( 1, 0, 0 )
zprimedirection= ( 0, 0, 1 )
range= ( 0, 360 )
clear
point= ( -RADIUS, 0 )
arc, radius= RADIUS, type= clockwise, size= small
point= ( RADIUS, 0)
doit
enddo
enddo
enddo
#
# the connecting rods in x-direction
#
do iz= 0, 1, 1
do iy= 0, 1, 1
-gccylinder
material= M3
radius= 0.1*LATTICE_D
length= INF
origin= ( -INF/2, \
iy*LATTICE_D, \
iz*LATTICE_D )
direction= ( 1, 0, 0 )
doit
enddo
enddo
#
# the connecting rods in y-direction
#
do iz= 0, 1, 1
do ix= 0, 1, 1
-gccylinder
material= M3
radius= 0.1*LATTICE_D
length= INF
origin= ( ix*LATTICE_D, \
-INF/2, \
iz*LATTICE_D )
direction= ( 0, 1, 0 )
doit
enddo
enddo
#
# the connecting rods in z-direction
#
do ix= 0, 1, 1
do iy= 0, 1, 1
-gccylinder
material= M3
radius= 0.1*LATTICE_D
length= INF
origin= ( ix*LATTICE_D, \
iy*LATTICE_D, \
-INF/2 )
direction= ( 0, 0, 1 )
doit
enddo
enddo
#
# definition of the material properties
#
-material
material= M3, type= electric
#
# what does the materialdistribution look like?
#
-volumeplot
## doit
#################
#
# computation of the eigenvalues
#
-eigenvalues
solutions= 20
estimation= 2.8
pfac2= 1e-2
passes= 2
doit
end
In order to compute the four parts of the Brillouin diagram, we use a
shell-script.
This shell script starts a program four times. That program starts gd1
several times to compute the frequencies for different phase shifts.
This is the shell-script:
#!/bin/sh
#
# Compile the program which starts "gd1" several times
#
f77 brillo.f -o brillo.a.out
for part in 1 2 3 4
do
#
# (re)create the file that defines which part of the
# Brillouin diagram is to be computed:
#
echo "define(PART, $part)" > this-part-of-brillo
#
# compute..
./brillo.a.out
#
# save the result, and display
#
cp brillo.mtv brillo.part=$part.mtv
mymtv brillo.part=$part.mtv &
done
#
# compile the program that combines the four parts
# to a single Brillouin diagram of a 3D structure,
# execute it,
# and display the result..
#
f90 a3dbrillo.f
cat brillo.part=[1-4].mtv | a.out
mymtv2 3D-brillo.mtv &
The following is the source of the program that starts gd1 several times to compute the frequencies for different phase shifts:
!
! /usr/local/gd1/examples-from-the-manual/brillo.f90
!
PROGRAM Bla
IMPLICIT NONE
CHARACTER(LEN= 400) cmd
REAL, DIMENSION(300,1000) :: f0
REAL, DIMENSION(1000) :: ph0
INTEGER :: i11, i19, NMode, np, ip, Mode, iDum
REAL :: af, ap, p0, p1, Phase, Acc, f
i11= 11
i19= 19
OPEN (UNIT= i19, &
FILE= 'brillo.mtv')
WRITE (UNIT= i19, FMT= 90)
90 FORMAT( &
'$ DATA= CURVE2D NAME= "Brillouin-Diagramm"',/, &
'% linetype= 0',/, &
'% markertype= 3',/, &
'% equalscale= false',/, &
'% fitpage= false',/, &
'% xyratio= 3',/, &
'% xlabel= "Phase-shift"',/, &
'% ylabel= "Frequency"',/, &
'% comment= "no comment"',/ )
af= 0
ap= 0
NMode= 15
! p0: First Phase
! p1: Last Phase
! np: Number of Phases
p0= 0.
p1= 180.
np= 41
DO ip= 1, np, 1
Phase= p0+(ip-1)*(p1-p0)/FLOAT(np-1)
ph0(ip)= Phase
WRITE (UNIT= cmd, FMT= 81) Phase
81 FORMAT( &
' gd1 "-DPHASE=', F8.2, '"< brillo.gdf ', &
'| tee brillo.tmp | grep "for me" > brillo.out' )
write (UNIT= 0, FMT= '(1X,4711(A))') ' cmd:', TRIM(cmd)
CALL system( cmd )
OPEN (UNIT= i11, &
FILE= 'brillo.out')
Mode= 0
100 CONTINUE
DO
READ (UNIT= i11, FMT= *, ERR= 199, END= 199) iDum, f, acc
IF (acc < 0.5) THEN
Mode= Mode+1
IF (Mode <= NMode) f0(Mode,ip)= f
WRITE (UNIT= i19, FMT= 71) Phase,f
write (0,*) Phase, f, acc
af= MAX(af, f)
ap= MAX(ap, Phase)
END IF
END DO
199 CONTINUE
CLOSE (UNIT= i11)
END DO
71 FORMAT( '@ point x1=', 1P, E12.6, ' y1=', E12.6, &
' z1= 0 markertype=1', ' markersize= 1' )
DO Mode= 1, NMode, 1
WRITE (UNIT= i19, FMT= *)
DO ip= 1, np, 1
WRITE (UNIT= i19, FMT= *) ph0(ip),f0(Mode,ip)
END DO
END DO
WRITE (UNIT= i19, FMT= *)
WRITE (UNIT= i19, FMT= 99) 0., 0., 0., af, ap, af
99 FORMAT( 3(1H ,1P, 2(E12.6, ' '), /), /)
!
! **
! ** write the uninterpreted data
! **
!
DO ip= 1, np, 1
WRITE (UNIT= i19, FMT= '(//,A,F14.2)') ' # Phase:', ph0(ip)
DO Mode= 1, NMode, 1
WRITE (UNIT= i19, FMT= '(A,1X,1P,E20.7)') '#', f0(Mode,ip)
END DO
END DO
END PROGRAM Bla
The resulting plots are presented in figure 5.9.
The following is the source of the program that combines the four parts of the Brillouin diagram into a single plot:
*
* /usr/local/gd1/examples-from-the-manual/a3dbrillo.f
*
* usage:
*
* cat brillo.part=[1-4].mtv | a.out
* mymtv2 3D-brillo.mtv
*
*
DIMENSION f0(300,1 000), ph0(1 000)
REAL, DIMENSION(100) :: Phase0, scale
CHARACTER(LEN=1000) :: str
*
i11= 11
i19= 19
OPEN (UNIT= i19
1 , FILE= '3D-brillo.mtv')
*
WRITE (UNIT= i19, FMT= 9000)
9000 FORMAT(
1 '$ DATA= CURVE2D NAME= "Brillouin-Diagramm"',/,
2 '% linetype= 0',/,
3 '% markertype= 3',/
4 '% equalscale= false',/,
5 '% fitpage= false',/,
6 '% xyratio= 3',/,
7 '% xlabel= "normalised Phase-shift"',/,
8 '% ylabel= "Frequency"',/,
9 '% comment= "no comment"',/
x )
*
af= 0.
ap= 0.
*
NMode= 10
*
phase0(1:4)= (/ 0.0,
1 1.0,
2 2.0,
3 4.0 /)
scale(1:4)= (/ 1./180.0, !! Gamma to H
2 1./180.0, !! H to N
3 1./180.0, !! N to P
4 -1./180.0 /) !! P to Gamma
*
Phase Last= 0.
np= 1
str= ' '
DO
DO
IF (INDEX(str, '# phase:') .NE. 0) THEN
jj= INDEX(str, '# phase:')+LEN('# phase:')
READ (UNIT= str(jj:), FMT= *) phase
IF (phase .LT. Phase Last) THEN
np= np+1
ENDIF
Phase Last= phase
EXIT
ENDIF
READ (UNIT= *, FMT= '(A)', END= 10) str
ENDDO
write (*,*) ' phase:', phase
pp= Phase0(np)
ph0(np)= pp+phase*scale(np)
*
mode= 0
DO mode= 1, NMode, 1
READ (UNIT= *, FMT= '(A)', END= 10) str
READ (UNIT= str(2:), FMT= *, IOSTAT= iostat) f
IF (iostat .NE. 0) EXIT
write (*,*) ' pp, f:', pp+phase*scale(np), f
f0(mode,np)= f
WRITE (UNIT= i19, FMT= 7010) pp+phase*scale(np),f
af= MAX(af,f)
ap= MAX(ap,pp+phase*scale(np))
ENDDO
ENDDO
10 CONTINUE
*
7010 FORMAT(
1 '@ point x1=',E12.6,' y1=',E12.6,' z1= 0 markertype=1',
2 ' markersize= 1')
*
WRITE (UNIT= i19, FMT= *)
WRITE (UNIT= i19, FMT= 99) 0.,0.,0.,af,ap,af
99 FORMAT(3(' ',2(E12.6,' ')/),/)
*
END
The resulting plot is presented in figure 5.10.
This is the end of this document